

Improved joint X-ray and neutron refinement procedure in Phenix
- PMID: 37942718
- PMCID: PMC10833352
- DOI: 10.1107/S2059798323008914
Improved joint X-ray and neutron refinement procedure in Phenix
Abstract
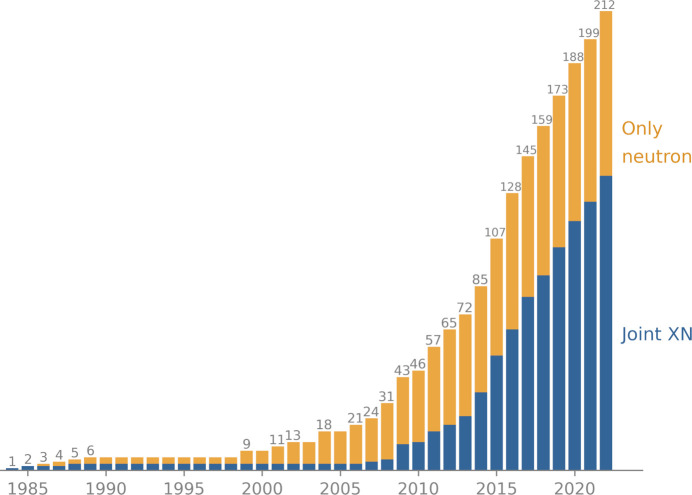
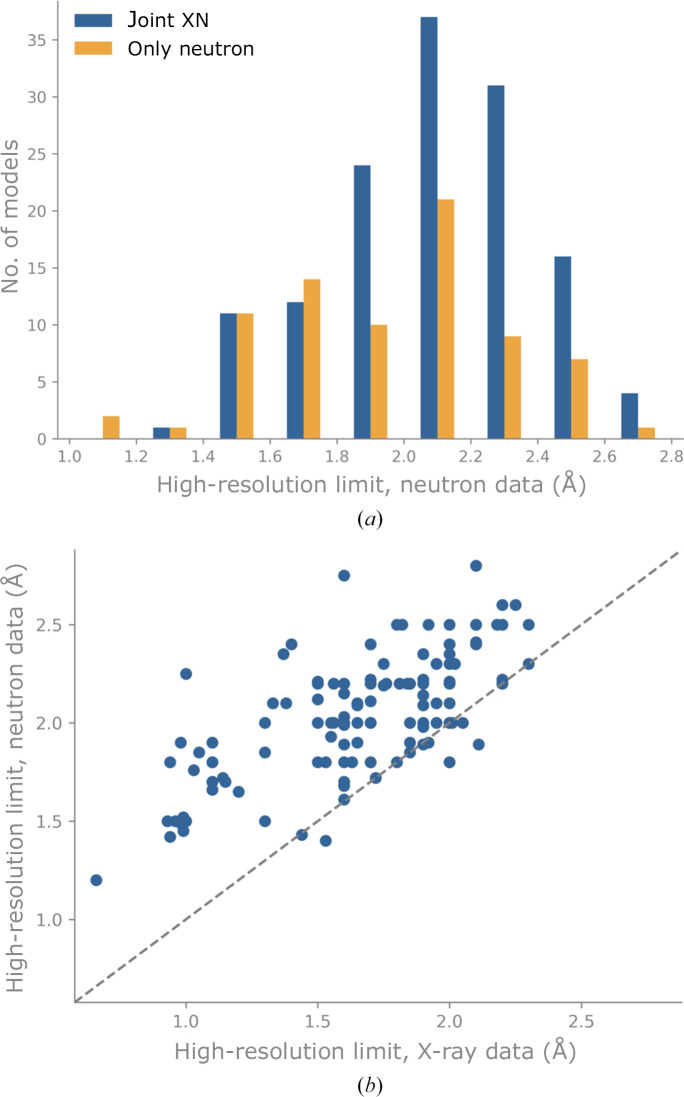
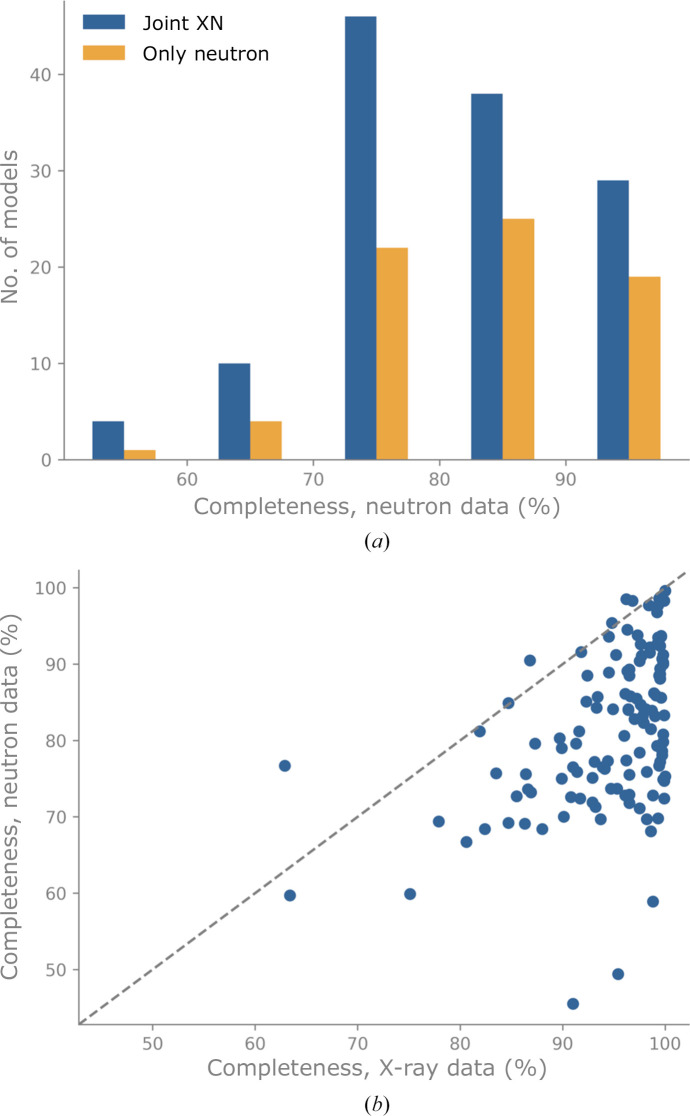
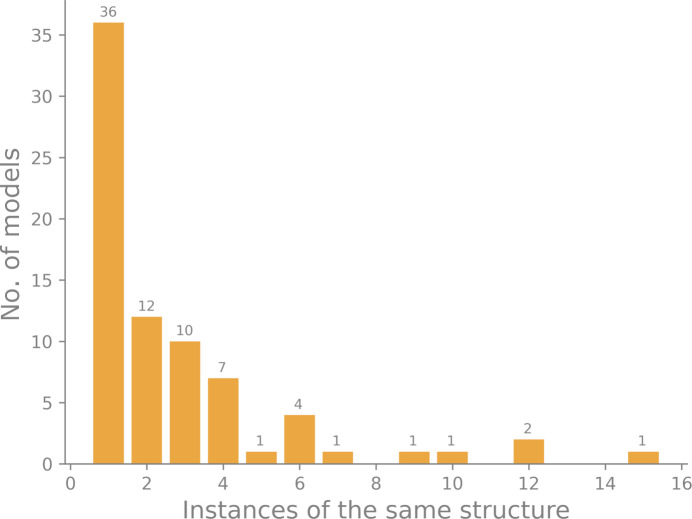
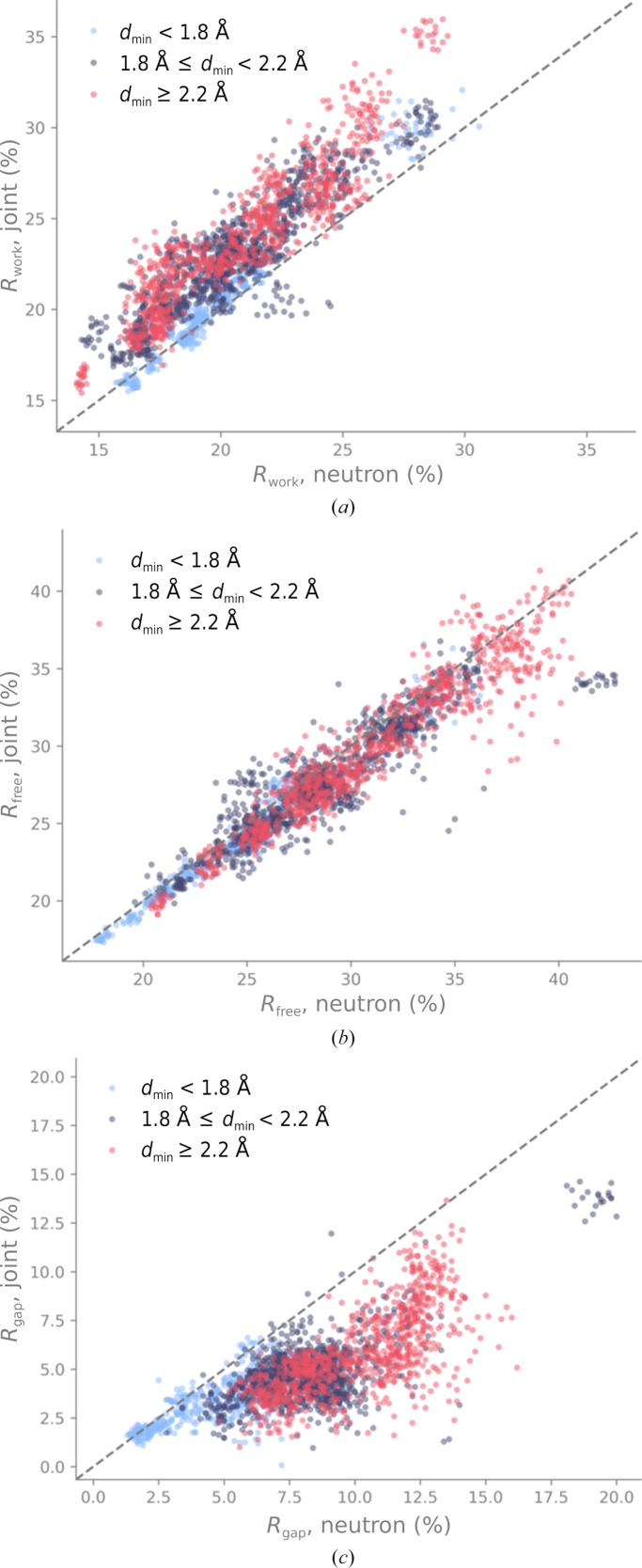
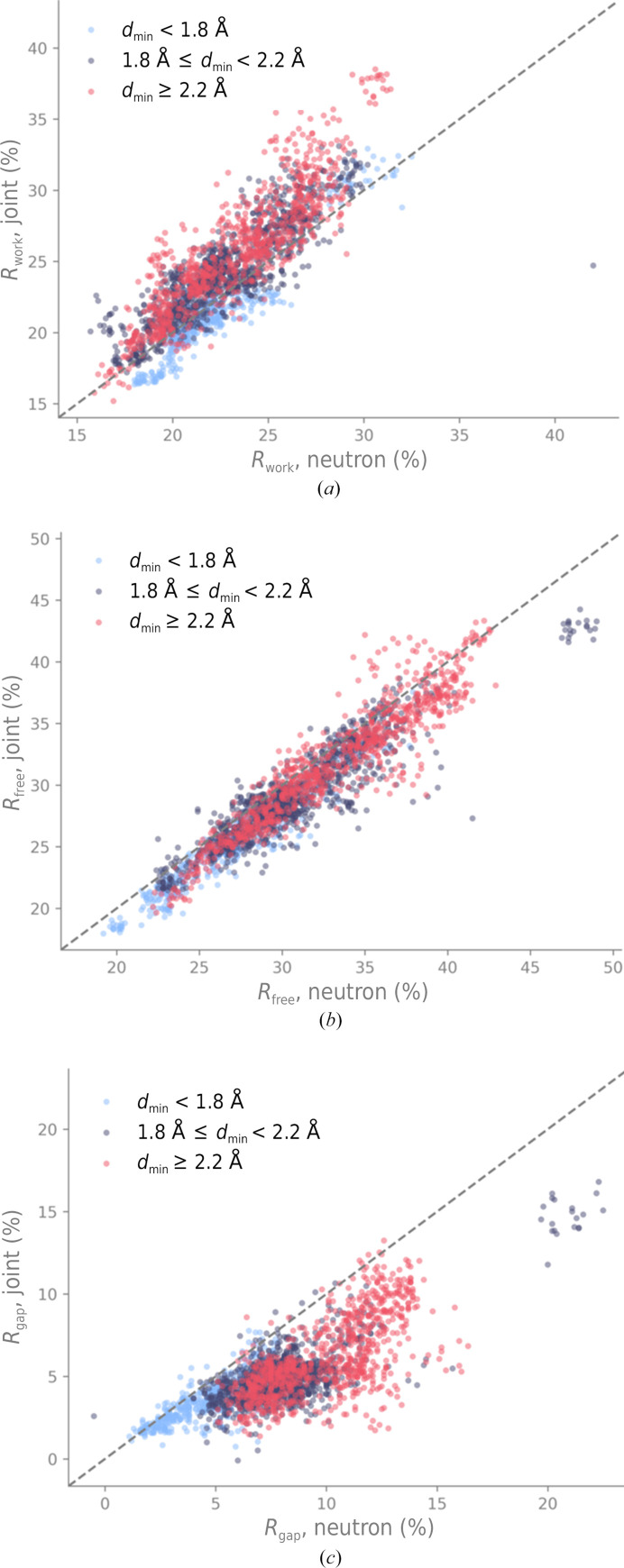
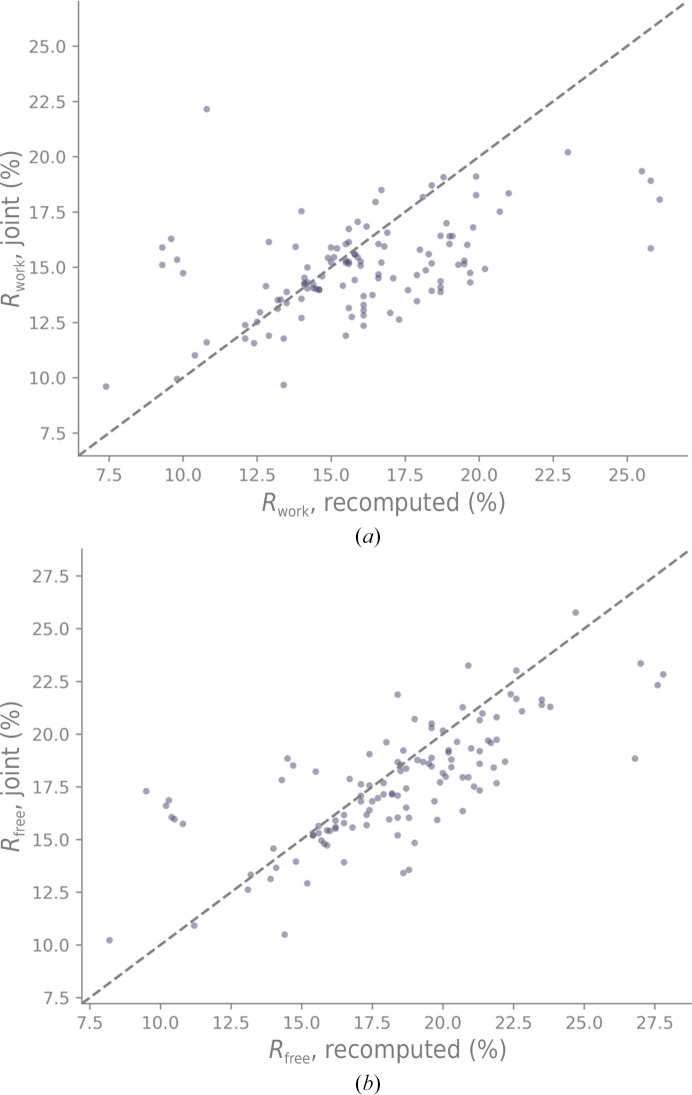
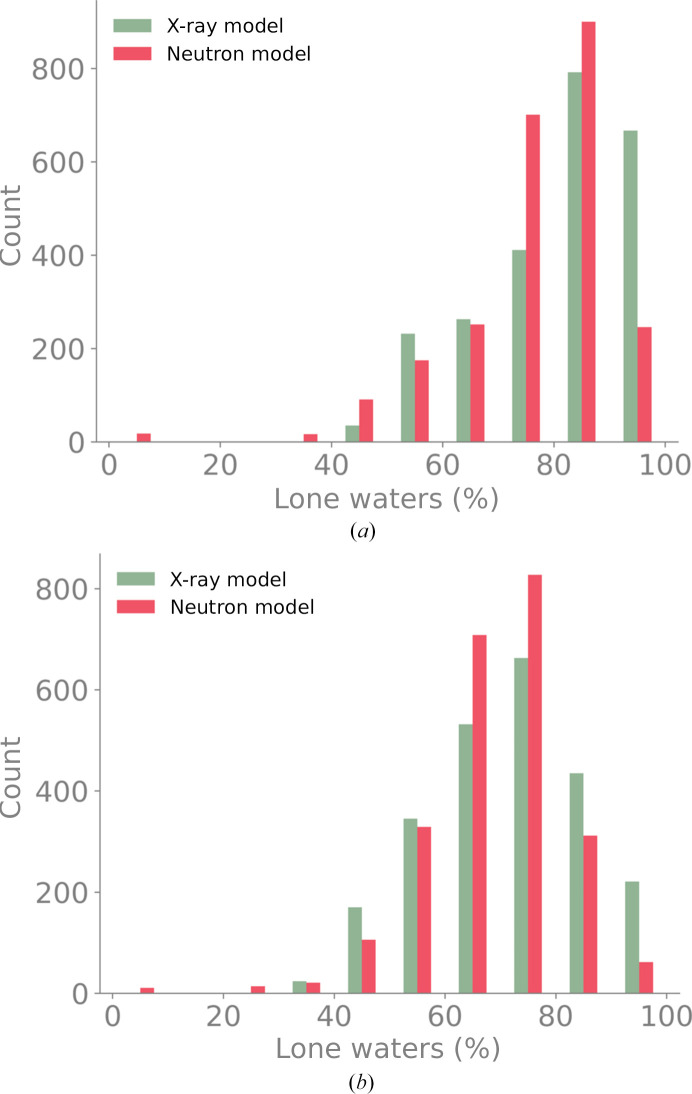
Neutron diffraction is one of the three crystallographic techniques (X-ray, neutron and electron diffraction) used to determine the atomic structures of molecules. Its particular strengths derive from the fact that H (and D) atoms are strong neutron scatterers, meaning that their positions, and thus protonation states, can be derived from crystallographic maps. However, because of technical limitations and experimental obstacles, the quality of neutron diffraction data is typically much poorer (completeness, resolution and signal to noise) than that of X-ray diffraction data for the same sample. Further, refinement is more complex as it usually requires additional parameters to describe the H (and D) atoms. The increase in the number of parameters may be mitigated by using the `riding hydrogen' refinement strategy, in which the positions of H atoms without a rotational degree of freedom are inferred from their neighboring heavy atoms. However, this does not address the issues related to poor data quality. Therefore, neutron structure determination often relies on the presence of an X-ray data set for joint X-ray and neutron (XN) refinement. In this approach, the X-ray data serve to compensate for the deficiencies of the neutron diffraction data by refining one model simultaneously against the X-ray and neutron data sets. To be applicable, it is assumed that both data sets are highly isomorphous, and preferably collected from the same crystals and at the same temperature. However, the approach has a number of limitations that are discussed in this work by comparing four separately re-refined neutron models. To address the limitations, a new method for joint XN refinement is introduced that optimizes two different models against the different data sets. This approach is tested using neutron models and data deposited in the Protein Data Bank. The efficacy of refining models with H atoms as riding or as individual atoms is also investigated.
Keywords: joint XN refinement; macromolecular crystallography; neutron diffraction.
open access.
Figures

Similar articles
-
What are the current limits on determination of protonation state using neutron macromolecular crystallography?Methods Enzymol. 2020;634:225-255. doi: 10.1016/bs.mie.2020.01.008. Epub 2020 Feb 13. Methods Enzymol. 2020. PMID: 32093835 Free PMC article.
-
Neutron crystallographic refinement with REFMAC5 from the CCP4 suite.Acta Crystallogr D Struct Biol. 2023 Dec 1;79(Pt 12):1056-1070. doi: 10.1107/S2059798323008793. Epub 2023 Nov 3. Acta Crystallogr D Struct Biol. 2023. PMID: 37921806 Free PMC article.
-
Implementation of the riding hydrogen model in CCTBX to support the next generation of X-ray and neutron joint refinement in Phenix.Methods Enzymol. 2020;634:177-199. doi: 10.1016/bs.mie.2020.01.007. Epub 2020 Feb 13. Methods Enzymol. 2020. PMID: 32093832 Free PMC article.
-
Evaluation of models determined by neutron diffraction and proposed improvements to their validation and deposition.Acta Crystallogr D Struct Biol. 2018 Aug 1;74(Pt 8):800-813. doi: 10.1107/S2059798318004588. Epub 2018 Jul 24. Acta Crystallogr D Struct Biol. 2018. PMID: 30082516 Free PMC article. Review.
-
Sub-atomic resolution X-ray crystallography and neutron crystallography: promise, challenges and potential.IUCrJ. 2015 Jun 30;2(Pt 4):464-74. doi: 10.1107/S2052252515011239. eCollection 2015 Jul 1. IUCrJ. 2015. PMID: 26175905 Free PMC article. Review.
Cited by
-
Quantum refinement in real and reciprocal space using the Phenix and ORCA software.IUCrJ. 2024 Nov 1;11(Pt 6):921-937. doi: 10.1107/S2052252524008406. IUCrJ. 2024. PMID: 39345101 Free PMC article.
References
-
- Afonine, P. V. (2015). Comput. Crystallogr. Newsl. 5, 37–46.
-
- Afonine, P. V. & Adams, P. D. (2012). Comput. Crystallogr. Newsl. 3, 18–21.
-
- Afonine, P. V., Echols, N., Grosse-Kunstleve, R. W., Moriarty, N. W. & Adams, P. D. (2011). Comput. Crystallogr. Newsl. 2, 99–103.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
