

Interplay between the DNA damage response and the life cycle of DNA tumor viruses
- PMID: 37918513
- PMCID: PMC10685005
- DOI: 10.1016/j.tvr.2023.200272
Interplay between the DNA damage response and the life cycle of DNA tumor viruses
Abstract
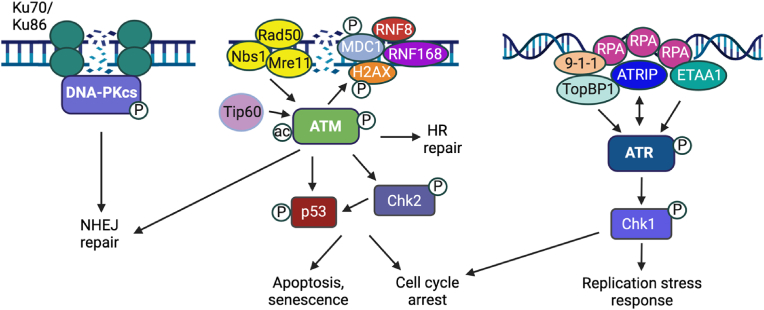
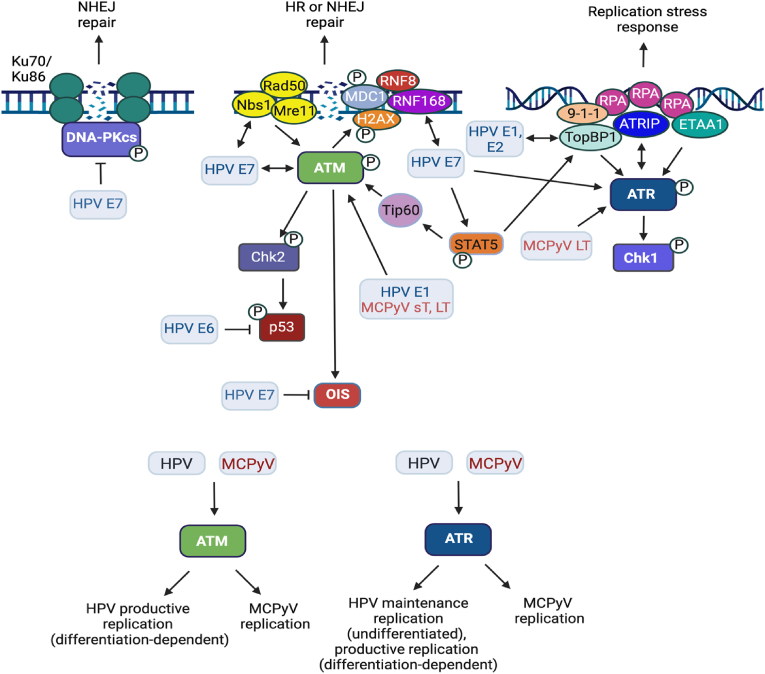
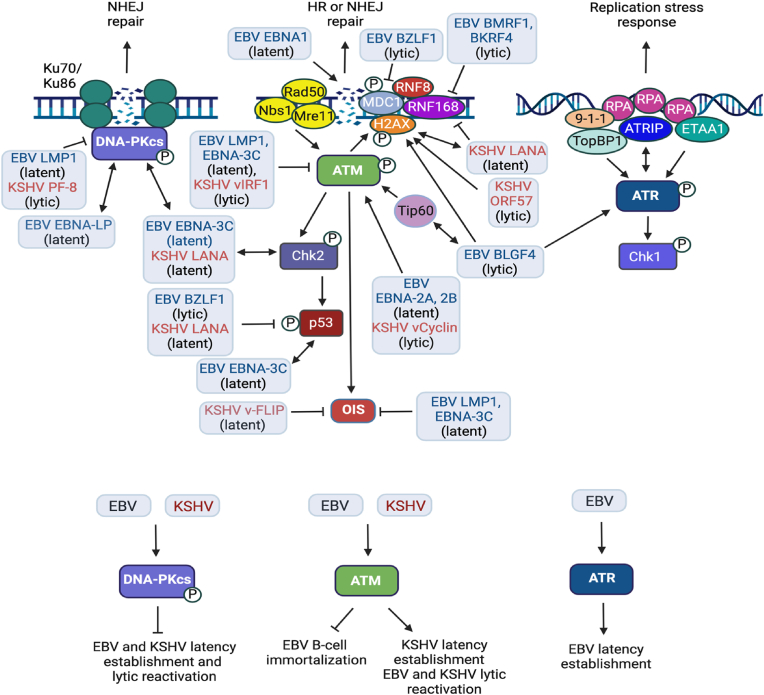
Approximately 20 % of human cancers are associated with virus infection. DNA tumor viruses can induce tumor formation in host cells by disrupting the cell's DNA replication and repair mechanisms. Specifically, these viruses interfere with the host cell's DNA damage response (DDR), which is a complex network of signaling pathways that is essential for maintaining the integrity of the genome. DNA tumor viruses can disrupt these pathways by expressing oncoproteins that mimic or inhibit various DDR components, thereby promoting genomic instability and tumorigenesis. Recent studies have highlighted the molecular mechanisms by which DNA tumor viruses interact with DDR components, as well as the ways in which these interactions contribute to viral replication and tumorigenesis. Understanding the interplay between DNA tumor viruses and the DDR pathway is critical for developing effective strategies to prevent and treat virally associated cancers. In this review, we discuss the current state of knowledge regarding the mechanisms by which human papillomavirus (HPV), merkel cell polyomavirus (MCPyV), Kaposi's sarcoma-associated herpesvirus (KSHV), and Epstein-Barr virus (EBV) interfere with DDR pathways to facilitate their respective life cycles, and the consequences of such interference on genomic stability and cancer development.
Copyright © 2023 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
Interplay Between KSHV and the Host DNA Damage Response.Front Cell Infect Microbiol. 2020 Dec 9;10:604351. doi: 10.3389/fcimb.2020.604351. eCollection 2020. Front Cell Infect Microbiol. 2020. PMID: 33425783 Free PMC article. Review.
-
Interplay between DNA tumor viruses and the host DNA damage response.Curr Top Microbiol Immunol. 2013;371:229-57. doi: 10.1007/978-3-642-37765-5_9. Curr Top Microbiol Immunol. 2013. PMID: 23686238 Free PMC article. Review.
-
Localization of Double-Strand Break Repair Proteins to Viral Replication Compartments following Lytic Reactivation of Kaposi's Sarcoma-Associated Herpesvirus.J Virol. 2017 Oct 27;91(22):e00930-17. doi: 10.1128/JVI.00930-17. Print 2017 Nov 15. J Virol. 2017. PMID: 28855246 Free PMC article.
-
A Screen for Epstein-Barr Virus Proteins That Inhibit the DNA Damage Response Reveals a Novel Histone Binding Protein.J Virol. 2018 Jun 29;92(14):e00262-18. doi: 10.1128/JVI.00262-18. Print 2018 Jul 15. J Virol. 2018. PMID: 29743367 Free PMC article.
-
Nuclear Innate Immune DNA Sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi's Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency.J Virol. 2016 Sep 12;90(19):8822-41. doi: 10.1128/JVI.01003-16. Print 2016 Oct 1. J Virol. 2016. PMID: 27466416 Free PMC article.
Cited by
-
Abortive Infection of Animal Cells: What Goes Wrong.Annu Rev Virol. 2024 Sep;11(1):193-213. doi: 10.1146/annurev-virology-100422-023037. Epub 2024 Aug 30. Annu Rev Virol. 2024. PMID: 38631917 Review.
-
The Possible Role of Pathogens and Chronic Immune Stimulation in the Development of Diffuse Large B-Cell Lymphoma.Biomedicines. 2024 Mar 14;12(3):648. doi: 10.3390/biomedicines12030648. Biomedicines. 2024. PMID: 38540261 Free PMC article. Review.
References
-
- Hustedt N., Durocher D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016;19(1):1–9. Epub 2016/12/23. doi: 10.1038/ncb3452. PubMed PMID: 28008184. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
