

The Frequency of Intermediate Alleles in Patients with Cerebellar Phenotypes
- PMID: 37906407
- PMCID: PMC11102406
- DOI: 10.1007/s12311-023-01620-7
The Frequency of Intermediate Alleles in Patients with Cerebellar Phenotypes
Abstract
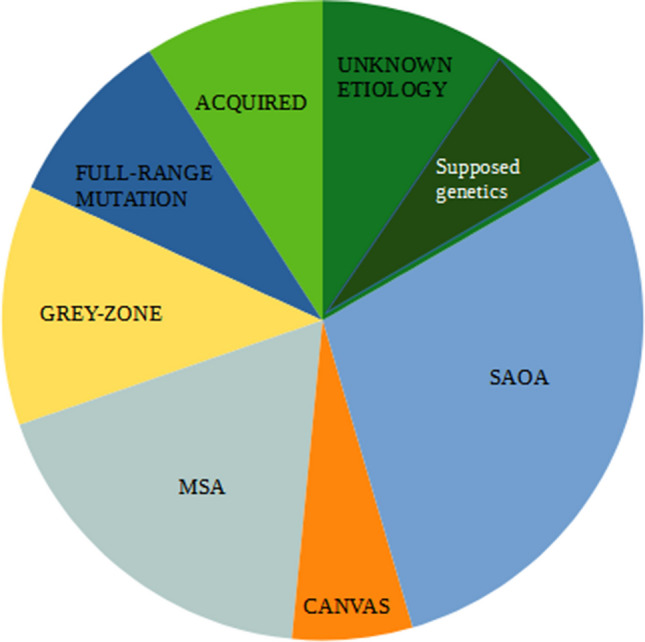
Cerebellar syndromes are clinically and etiologically heterogeneous and can be classified as hereditary, neurodegenerative non-hereditary, or acquired. Few data are available on the frequency of each form in the clinical setting. Growing interest is emerging regarding the genetic forms caused by triplet repeat expansions. Alleles with repeat expansion lower than the pathological threshold, termed intermediate alleles (IAs), have been found to be associated with disease manifestation. In order to assess the relevance of IAs as a cause of cerebellar syndromes, we enrolled 66 unrelated Italian ataxic patients and described the distribution of the different etiology of their syndromes and the frequency of IAs. Each patient underwent complete clinical, hematological, and neurophysiological assessments, neuroimaging evaluations, and genetic tests for autosomal dominant cerebellar ataxia (SCA) and fragile X-associated tremor/ataxia syndrome (FXTAS). We identified the following diagnostic categories: 28% sporadic adult-onset ataxia, 18% cerebellar variant of multiple system atrophy, 9% acquired forms, 9% genetic forms with full-range expansion, and 12% cases with intermediate-range expansion. The IAs were six in the FMR1 gene, two in the gene responsible for SCA8, and one in the ATXN2 gene. The clinical phenotype of patients carrying the IAs resembles, in most of the cases, the one associated with full-range expansion. Our study provides an exhaustive description of the causes of cerebellar ataxia, estimating for the first time the frequency of IAs in SCAs- and FXTAS-associated genes. The high percentage of cases with IAs supports further screening among patients with cerebellar syndromes.
Keywords: Cerebellar ataxia; FXTAS; Gray zone; Intermediate allele; SCA2; SCA8.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Frequency of FMR1 Premutation Alleles in Patients with Undiagnosed Cerebellar Ataxia and Multiple System Atrophy in the Japanese Population.Cerebellum. 2022 Dec;21(6):954-962. doi: 10.1007/s12311-021-01329-5. Epub 2021 Nov 29. Cerebellum. 2022. PMID: 34845661
-
Fragile X-associated tremor/ataxia syndrome: influence of the FMR1 gene on motor fiber tracts in males with normal and premutation alleles.JAMA Neurol. 2013 Aug;70(8):1022-9. doi: 10.1001/jamaneurol.2013.2934. JAMA Neurol. 2013. PMID: 23753897 Free PMC article.
-
Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS).Neurology. 2007 Aug 28;69(9):851-9. doi: 10.1212/01.wnl.0000269781.10417.7b. Neurology. 2007. PMID: 17724287
-
Unstable mutations in the FMR1 gene and the phenotypes.Adv Exp Med Biol. 2012;769:78-114. doi: 10.1007/978-1-4614-5434-2_6. Adv Exp Med Biol. 2012. PMID: 23560306 Free PMC article. Review.
-
Fragile X-associated tremor/ataxia syndrome: phenotypic comparisons with other movement disorders.Clin Neuropsychol. 2016 Aug;30(6):849-900. doi: 10.1080/13854046.2016.1202239. Clin Neuropsychol. 2016. PMID: 27414076 Free PMC article. Review.
Cited by
-
Early-onset phenotype in a patient with an intermediate allele and a large SCA1 expansion: a case report.BMC Neurol. 2024 Sep 17;24(1):348. doi: 10.1186/s12883-024-03846-2. BMC Neurol. 2024. PMID: 39289638 Free PMC article.
References
-
- Mongelli A, Magri S, Salvatore E, Rizzo E, De Rosa A, Fico T, Gatti M, Gellera C, Taroni F, Mariotti C, Nanetti L. Frequency and distribution of polyQ disease intermediate-length repeat alleles in healthy Italian population. Neurol Sci. 2020;41(6):1475–1482. doi: 10.1007/s10072-019-04233-3. - DOI - PubMed
-
- Gardiner SL, Boogaard MW, Trompet S, de Mutsert R, Rosendaal FR, Gussekloo J, Jukema JW, Roos RAC, Aziz NA. Prevalence of carriers of intermediate and pathological polyglutamine disease-associated alleles among large population-based cohorts. JAMA Neurol. 2019;76(6):650–656. doi: 10.1001/jamaneurol.2019.0423. - DOI - PMC - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
