

Detection and characterization of novel luchacoviruses, genus Alphacoronavirus, in saliva and feces of meso-carnivores in the northeastern United States
- PMID: 37882520
- PMCID: PMC10688340
- DOI: 10.1128/jvi.00829-23
Detection and characterization of novel luchacoviruses, genus Alphacoronavirus, in saliva and feces of meso-carnivores in the northeastern United States
Abstract
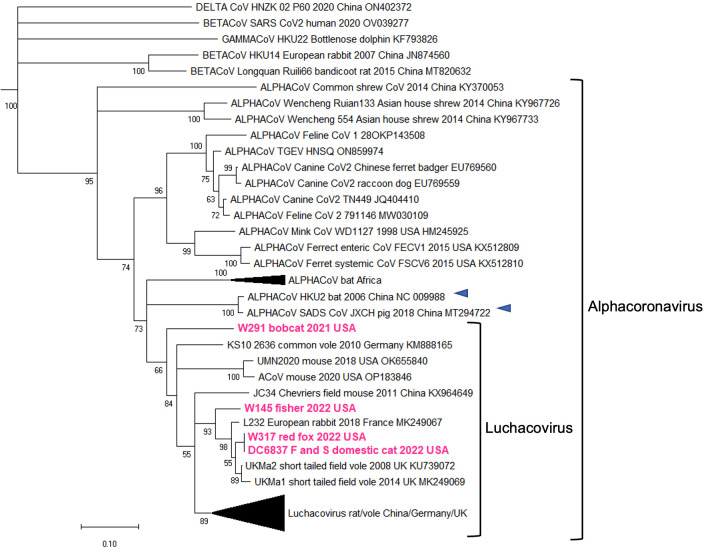
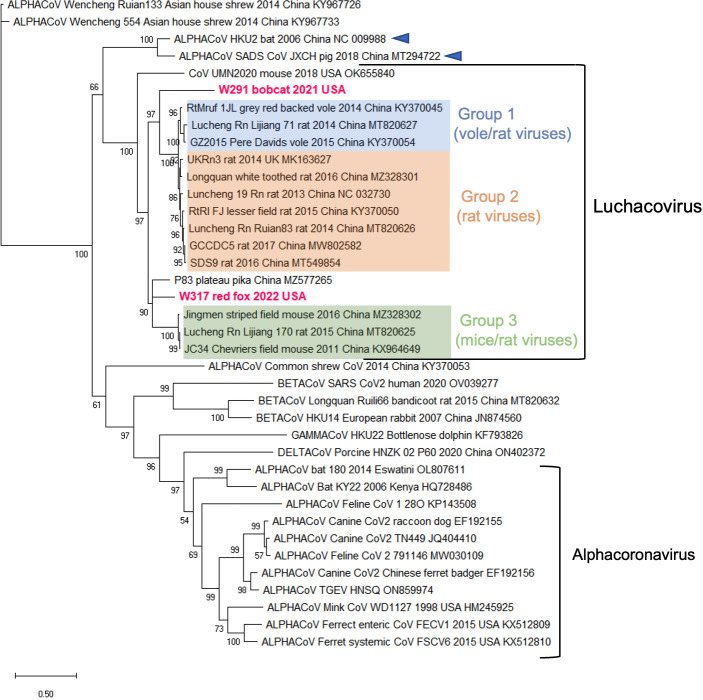
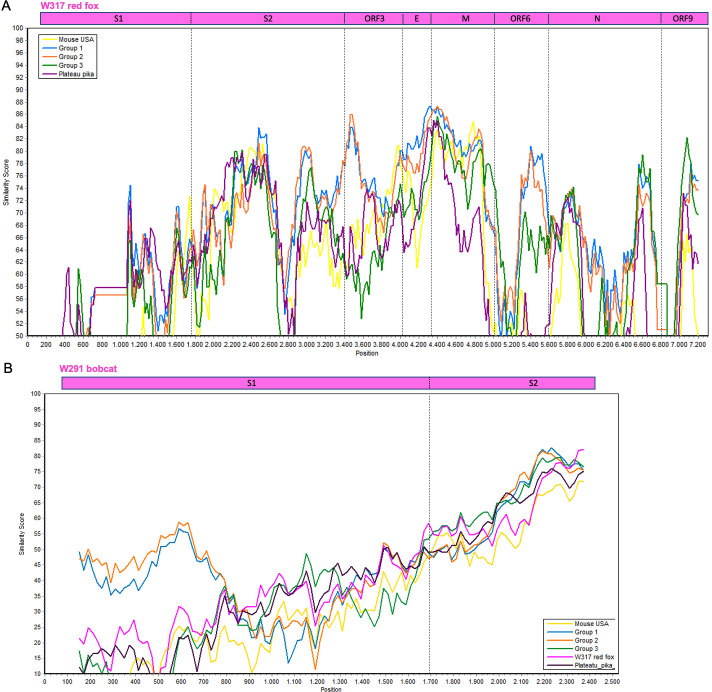
Several coronaviruses (CoVs) have been detected in domesticated, farmed, and wild meso-carnivores, causing a wide range of diseases and infecting diverse species, highlighting their important but understudied role in the epidemiology of these viruses. Assessing the viral diversity hosted in wildlife species is essential to understand their significance in the cross-species transmission of CoVs. Our focus here was on CoV discovery in meso-carnivores in the Northeast United States as a potential "hotspot" area with high density of humans and urban wildlife. This study identifies novel alphacoronaviruses circulating in multiple free-ranging wild and domestic species in this area and explores their potential epidemiological importance based on regions of the Spike gene, which are relevant for virus-host interactions.
Keywords: coronavirus; genomics; surveillance studies; wildlife.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Update of
-
Detection and characterization of novel luchacoviruses, genus Alphacoronavirus, shed in saliva and feces of meso-carnivores in the northeastern United States.bioRxiv [Preprint]. 2023 Sep 13:2023.05.31.541188. doi: 10.1101/2023.05.31.541188. bioRxiv. 2023. Update in: J Virol. 2023 Nov 30;97(11):e0082923. doi: 10.1128/jvi.00829-23 PMID: 37745528 Free PMC article. Updated. Preprint.
Similar articles
-
Detection and characterization of novel luchacoviruses, genus Alphacoronavirus, shed in saliva and feces of meso-carnivores in the northeastern United States.bioRxiv [Preprint]. 2023 Sep 13:2023.05.31.541188. doi: 10.1101/2023.05.31.541188. bioRxiv. 2023. Update in: J Virol. 2023 Nov 30;97(11):e0082923. doi: 10.1128/jvi.00829-23 PMID: 37745528 Free PMC article. Updated. Preprint.
-
Detection and Characterization of Distinct Alphacoronaviruses in Five Different Bat Species in Denmark.Viruses. 2018 Sep 11;10(9):486. doi: 10.3390/v10090486. Viruses. 2018. PMID: 30208582 Free PMC article.
-
Coronavirus genotype diversity and prevalence of infection in wild carnivores in the Serengeti National Park, Tanzania.Arch Virol. 2013 Apr;158(4):729-34. doi: 10.1007/s00705-012-1562-x. Epub 2012 Dec 5. Arch Virol. 2013. PMID: 23212740 Free PMC article.
-
Animal coronaviruses: what can they teach us about the severe acute respiratory syndrome?Rev Sci Tech. 2004 Aug;23(2):643-60. doi: 10.20506/rst.23.2.1513. Rev Sci Tech. 2004. PMID: 15702725 Review.
-
Bat-Origin Coronaviruses Expand Their Host Range to Pigs.Trends Microbiol. 2018 Jun;26(6):466-470. doi: 10.1016/j.tim.2018.03.001. Epub 2018 Apr 18. Trends Microbiol. 2018. PMID: 29680361 Free PMC article. Review.
References
-
- Siddell SG, Walker PJ, Lefkowitz EJ, Mushegian AR, Dempsey DM, Dutilh BE, Harrach B, Harrison RL, Hendrickson RC, Junglen S, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Nibert M, Rubino L, Sabanadzovic S, Simmonds P, Varsani A, Zerbini FM, Davison AJ. 2019. Additional changes to taxonomy ratified in a special vote by the international committee on taxonomy of viruses (October 2018). Arch Virol 164:2417–2429. doi:10.1007/s00705-019-04306-w - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
