

Repurposing of known drugs for COVID-19 using molecular docking and simulation analysis
- PMID: 37814677
- PMCID: PMC10560309
- DOI: 10.6026/97320630019149
Repurposing of known drugs for COVID-19 using molecular docking and simulation analysis
Abstract
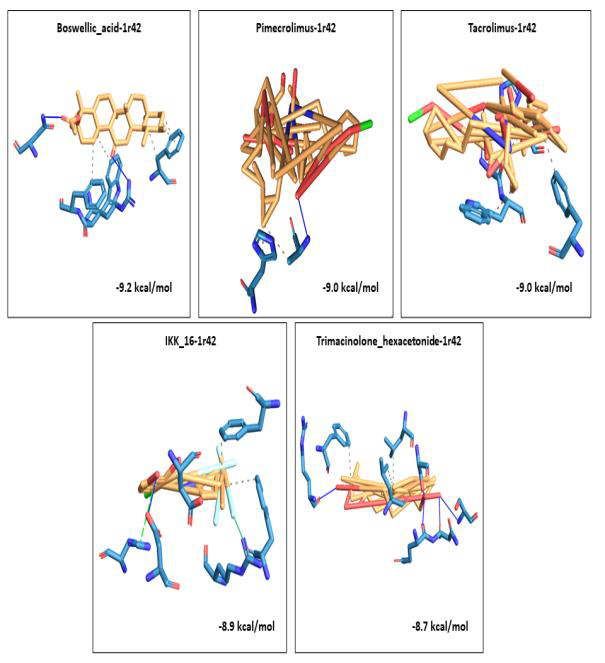
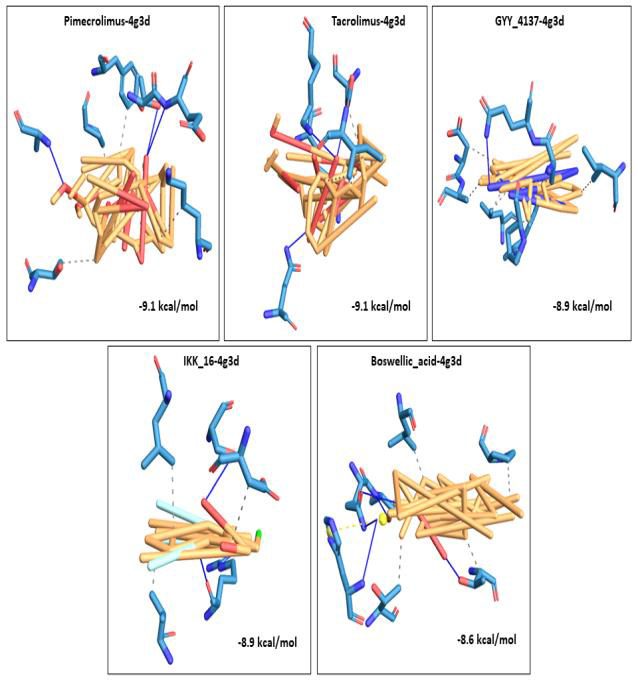
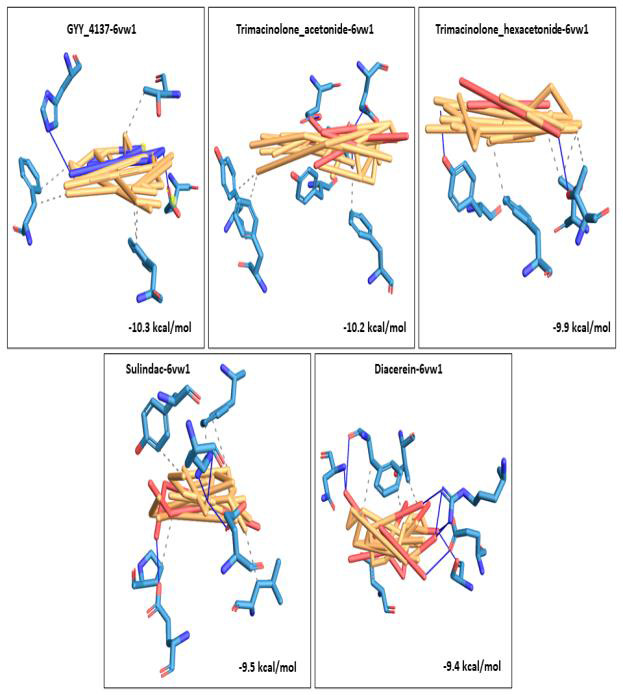
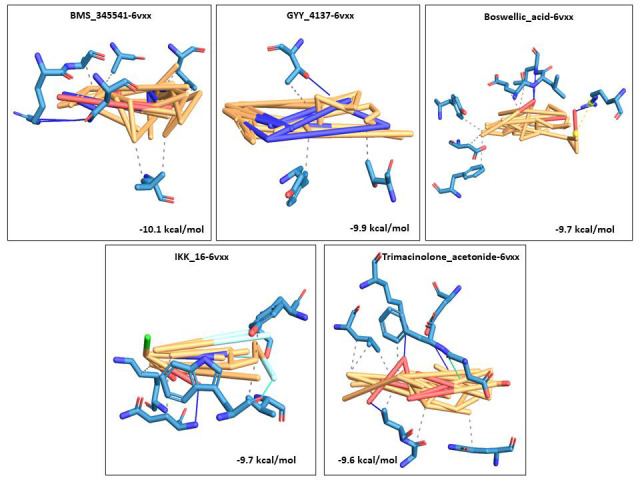
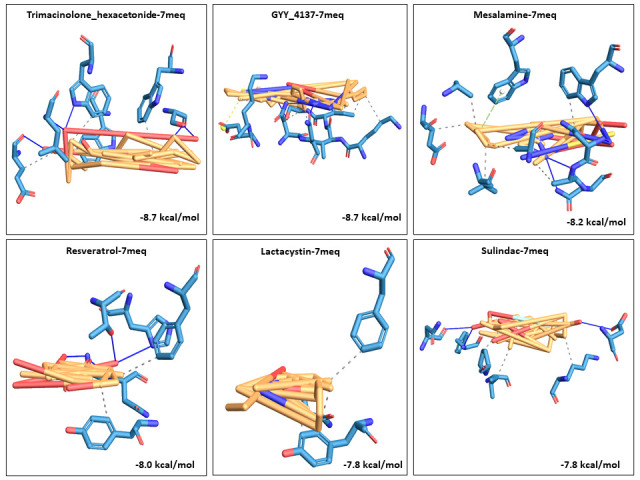
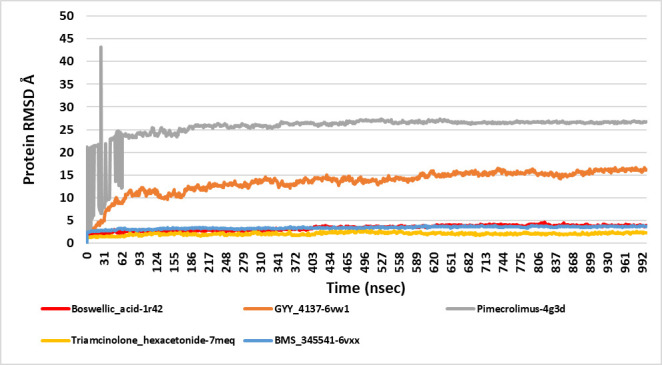
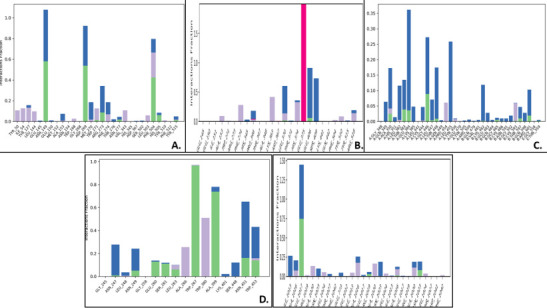
We selected fifty one drugs already known for their potential disease treatment roles in various studies and subjected to docking and molecular docking simulation (MDS) analyses. Five of them showed promising features that are discussed and suggested as potential candidates for repurposing for COVID-19. These top five compounds were boswellic acid, pimecrolimus, GYY-4137, BMS-345541 and triamcinolone hexacetonide that interacted with the chosen receptors 1R42, 4G3D, 6VW1, 6VXX and 7MEQ, respectively with binding energies of -9.2 kcal/mol, -9.1 kcal/mol, -10.3 kcal/mol, -10.1 kcal/mol and -8.7 kcal/mol, respectively. The MDS studies for the top 5 best complexes revealed binding features for the chosen receptor, human NF-kappa B transcription factor as an important drug target in COVID-19-based drug development strategies.
Keywords: COVID-19; SARS-CoV-2; ligand; molecular dynamics simulation; receptor.
© 2023 Biomedical Informatics.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

Similar articles
-
Repurposing immune boosting and anti-viral efficacy of Parkia bioactive entities as multi-target directed therapeutic approach for SARS-CoV-2: exploration of lead drugs by drug likeness, molecular docking and molecular dynamics simulation methods.J Biomol Struct Dyn. 2024 Jan-Feb;42(1):43-81. doi: 10.1080/07391102.2023.2192797. Epub 2023 Apr 5. J Biomol Struct Dyn. 2024. PMID: 37021347
-
Repurposing drugs and identification of inhibitors of integral proteins (spike protein and main protease) of SARS-CoV-2.J Biomol Struct Dyn. 2022 Sep;40(14):6587-6602. doi: 10.1080/07391102.2021.1886993. Epub 2021 Feb 16. J Biomol Struct Dyn. 2022. PMID: 33590806 Free PMC article.
-
Virtual Screening of Artemisia annua Phytochemicals as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme.Molecules. 2022 Nov 21;27(22):8103. doi: 10.3390/molecules27228103. Molecules. 2022. PMID: 36432204 Free PMC article.
-
In silico molecular docking, dynamics simulation and repurposing of some VEGFR-2 inhibitors based on the SARS-CoV-2-main-protease inhibitor N3.J Biomol Struct Dyn. 2023 Nov;41(19):9267-9281. doi: 10.1080/07391102.2022.2148000. Epub 2022 Nov 18. J Biomol Struct Dyn. 2023. PMID: 36399002 Review.
-
Phytocompounds of Rheum emodi, Thymus serpyllum, and Artemisia annua Inhibit Spike Protein of SARS-CoV-2 Binding to ACE2 Receptor: In Silico Approach.Curr Pharmacol Rep. 2021;7(4):135-149. doi: 10.1007/s40495-021-00259-4. Epub 2021 Jul 15. Curr Pharmacol Rep. 2021. PMID: 34306988 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous