

Bioactive lipid-based therapeutic approach to COVID-19 and other similar infections
- PMID: 37732033
- PMCID: PMC10507771
- DOI: 10.5114/aoms/135703
Bioactive lipid-based therapeutic approach to COVID-19 and other similar infections
Abstract
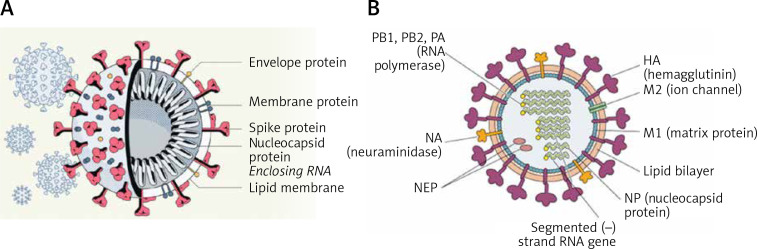
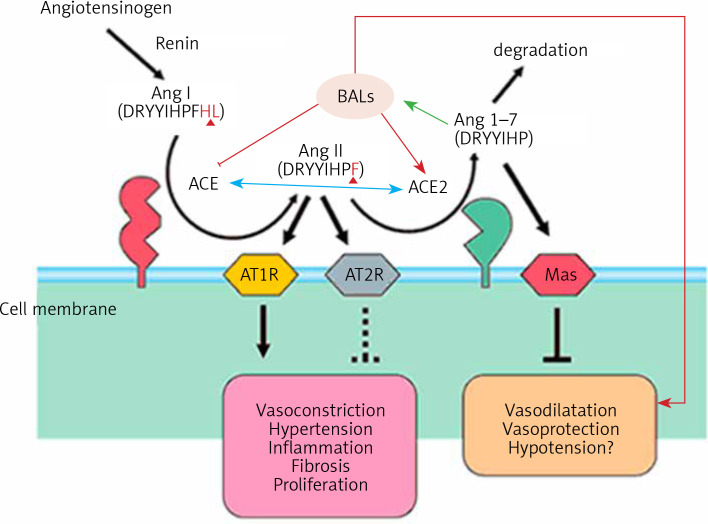
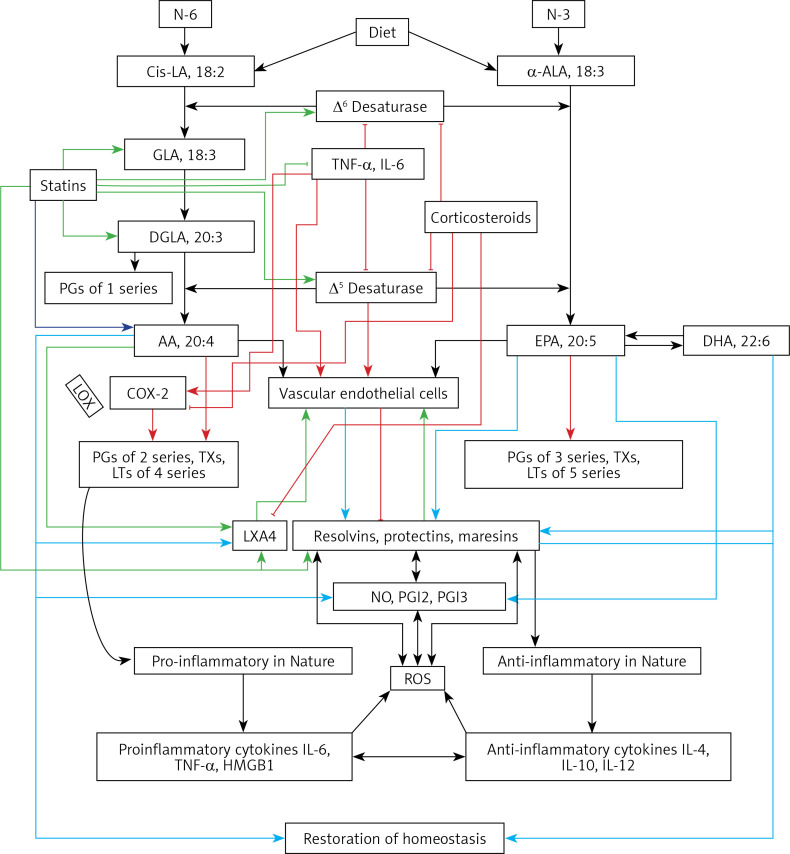
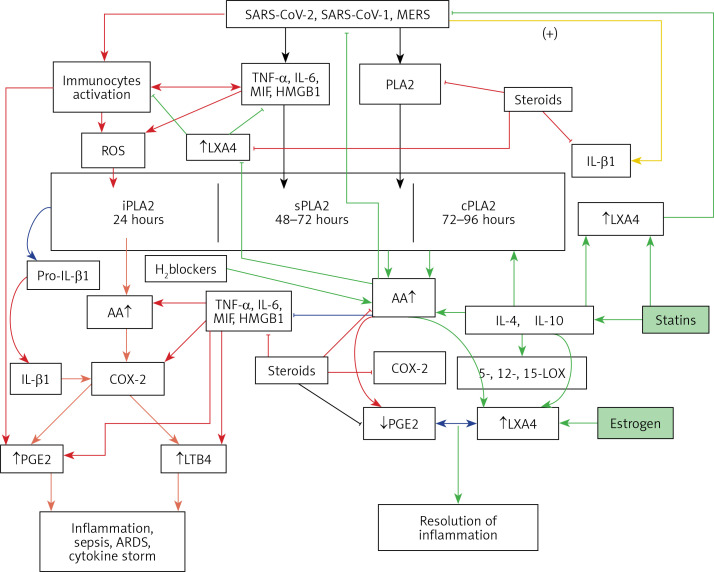
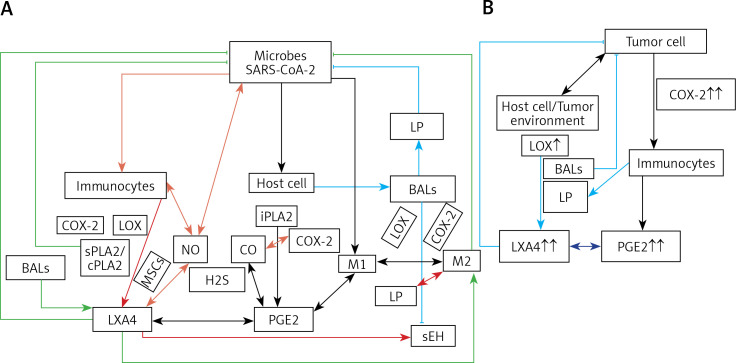
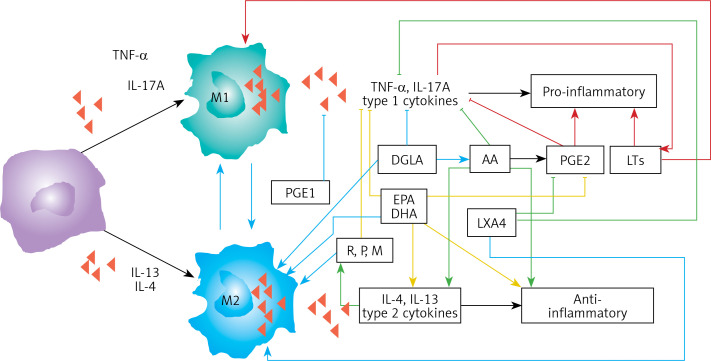
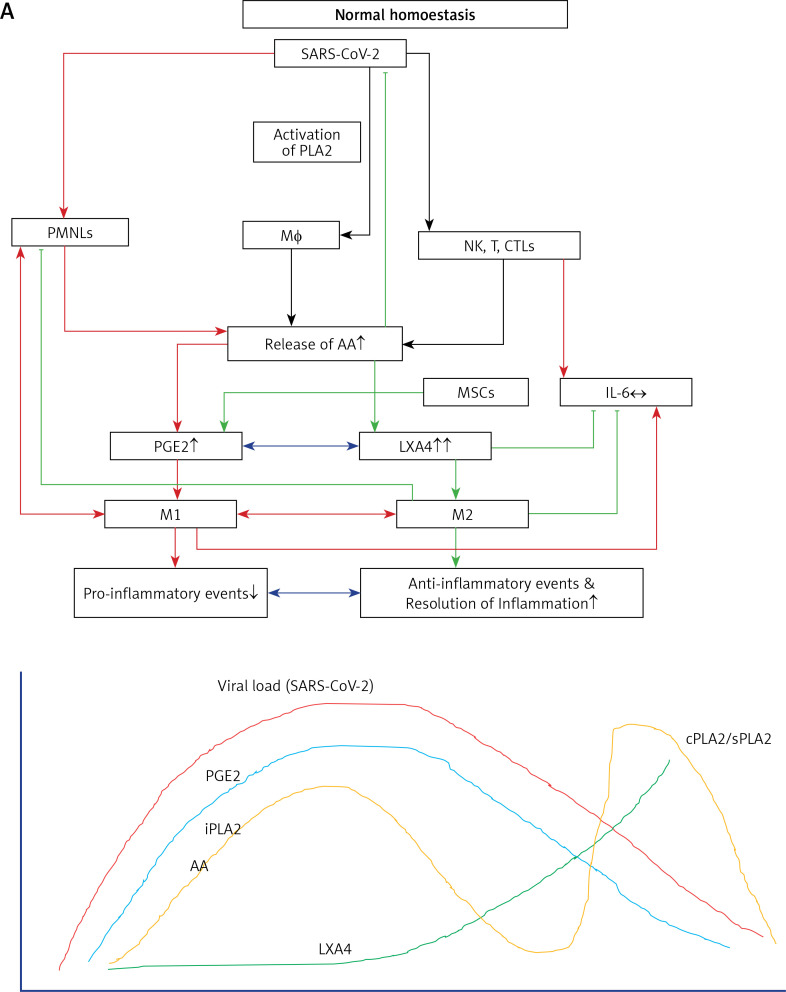
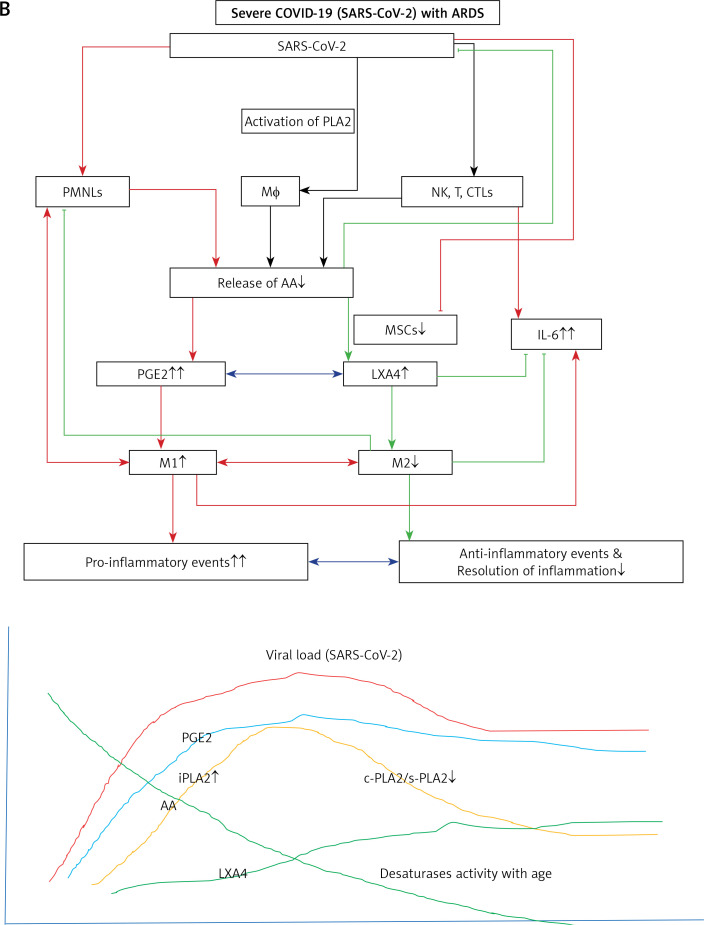
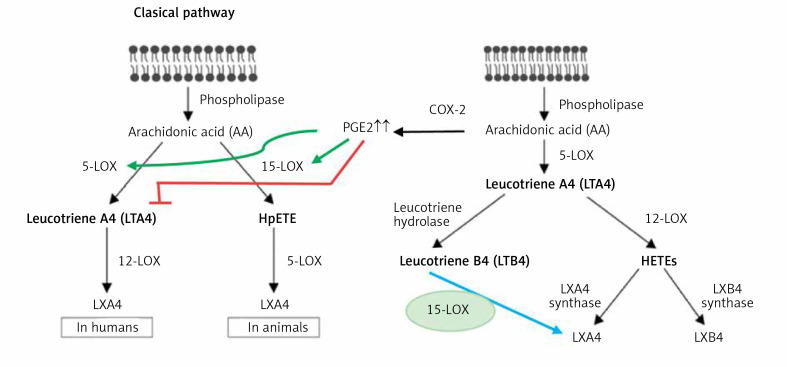
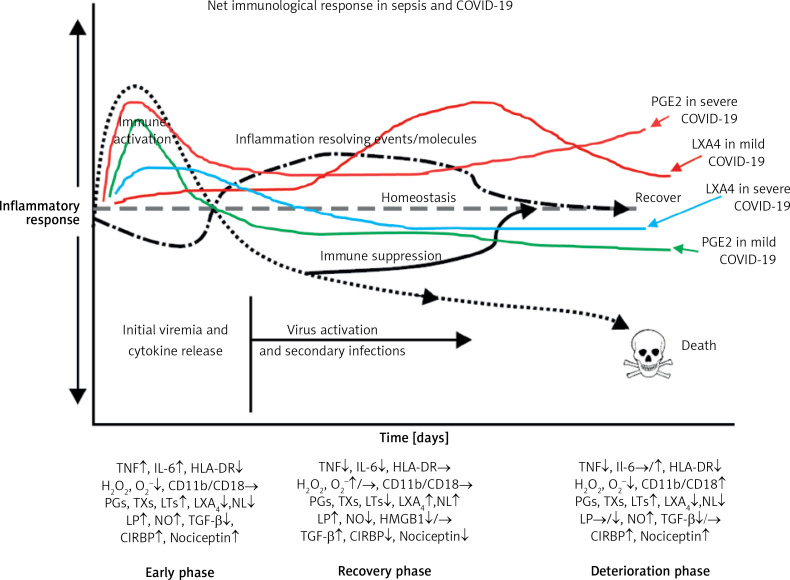
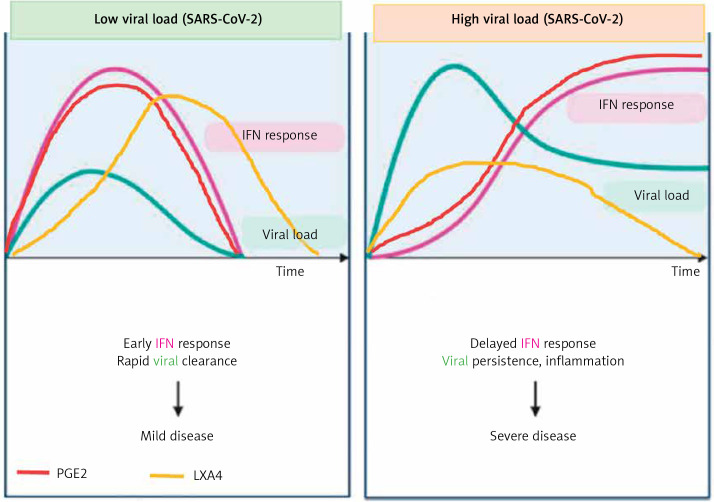
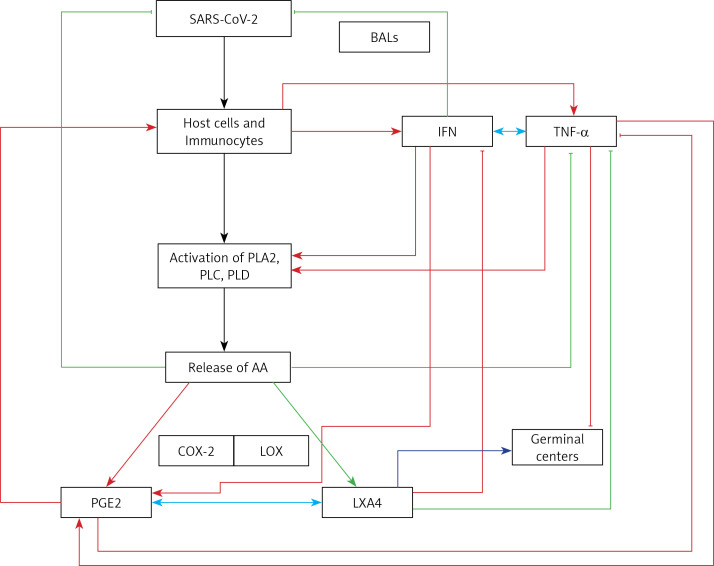
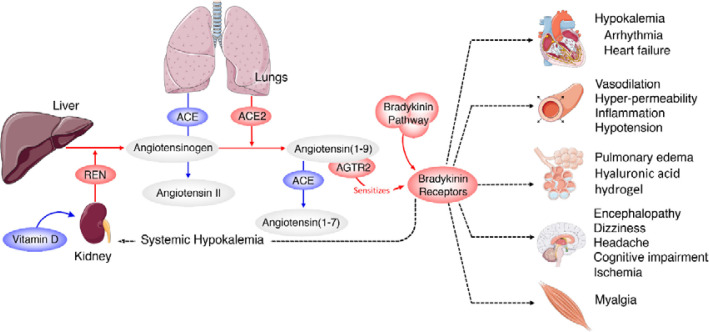
COVID-19 is caused by SARS-CoV-2 infection. Epithelial and T, NK, and other immunocytes release bioactive lipids especially arachidonic acid (AA) in response to microbial infections to inactivate them and upregulate the immune system. COVID-19 (coronavirus) and other enveloped viruses including severe acute respiratory syndrome (SARS-CoV-1 of 2002-2003) and Middle East respiratory syndrome (MERS; 2012-ongoing) and hepatitis B and C (HBV and HCV) can be inactivated by AA, γ-linolenic acid (GLA, dihomo-GLA (DGLA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), which are precursors to several eicosanoids. Prostaglandin E1, lipoxin A4, resolvins, protectins and maresins enhance phagocytosis of macrophages and leukocytes to clear debris from the site(s) of infection and injury, enhance microbial clearance and wound healing to restore homeostasis. Bioactive lipids modulate the generation of M1 and M2 macrophages and the activity of other immunocytes. Mesenchymal and adipose tissue-derived stem cells secrete LXA4 and other bioactive lipids to bring about their beneficial actions in COVID-19. Bioactive lipids regulate vasomotor tone, inflammation, thrombosis, immune response, inactivate enveloped viruses, regulate T cell proliferation and secretion of cytokines, stem cell survival, proliferation and differentiation, and leukocyte and macrophage functions, JAK kinase activity and neutrophil extracellular traps and thus, have a critical role in COVID-19.
Keywords: COVID-19; SARS-CoV-2; bioactive lipids; essential fatty acids; inflammation; lipoxin A4; macrophages; maresins; polyunsaturated fatty acids; prostaglandins; protectins; resolvins.
Copyright: © 2021 Termedia & Banach.
Conflict of interest statement
The author declares no conflict of interest.
Figures
Similar articles
-
Can Bioactive Lipids Inactivate Coronavirus (COVID-19)?Arch Med Res. 2020 Apr;51(3):282-286. doi: 10.1016/j.arcmed.2020.03.004. Epub 2020 Mar 27. Arch Med Res. 2020. PMID: 32229155 Free PMC article.
-
Bioactive Lipids in COVID-19-Further Evidence.Arch Med Res. 2021 Jan;52(1):107-120. doi: 10.1016/j.arcmed.2020.09.006. Epub 2020 Sep 9. Arch Med Res. 2021. PMID: 32981754 Free PMC article. Review.
-
"Cell Membrane Theory of Senescence" and the Role of Bioactive Lipids in Aging, and Aging Associated Diseases and Their Therapeutic Implications.Biomolecules. 2021 Feb 8;11(2):241. doi: 10.3390/biom11020241. Biomolecules. 2021. PMID: 33567774 Free PMC article. Review.
-
Arachidonic acid and other unsaturated fatty acids and some of their metabolites function as endogenous antimicrobial molecules: A review.J Adv Res. 2018 Jan 3;11:57-66. doi: 10.1016/j.jare.2018.01.001. eCollection 2018 May. J Adv Res. 2018. PMID: 30034876 Free PMC article. Review.
-
Essential Fatty Acids and Their Metabolites in the Pathobiology of Inflammation and Its Resolution.Biomolecules. 2021 Dec 14;11(12):1873. doi: 10.3390/biom11121873. Biomolecules. 2021. PMID: 34944517 Free PMC article. Review.
Cited by
-
The effect of polyunsaturated fatty acids on the severity and mortality of COVID patients: A systematic review.Life Sci. 2022 Jun 15;299:120489. doi: 10.1016/j.lfs.2022.120489. Epub 2022 Mar 28. Life Sci. 2022. PMID: 35358595 Free PMC article. Review.
-
Tailored lipopeptide surfactants as potentially effective drugs to treat SARS-CoV-2 infection.Med Hypotheses. 2022 Oct;167:110948. doi: 10.1016/j.mehy.2022.110948. Epub 2022 Sep 17. Med Hypotheses. 2022. PMID: 36157252 Free PMC article.
-
COVID-19 and lipids. The role of lipid disorders and statin use in the prognosis of patients with SARS-CoV-2 infection.Lipids Health Dis. 2021 Oct 25;20(1):141. doi: 10.1186/s12944-021-01563-0. Lipids Health Dis. 2021. PMID: 34689776 Free PMC article. Review.
-
Can essential fatty acids (EFAs) prevent and ameliorate post-COVID-19 long haul manifestations?Lipids Health Dis. 2024 Apr 19;23(1):112. doi: 10.1186/s12944-024-02090-4. Lipids Health Dis. 2024. PMID: 38641607 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous