

Incorporating physics to overcome data scarcity in predictive modeling of protein function: A case study of BK channels
- PMID: 37713443
- PMCID: PMC10529646
- DOI: 10.1371/journal.pcbi.1011460
Incorporating physics to overcome data scarcity in predictive modeling of protein function: A case study of BK channels
Abstract
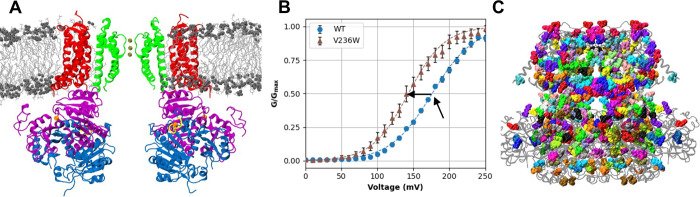
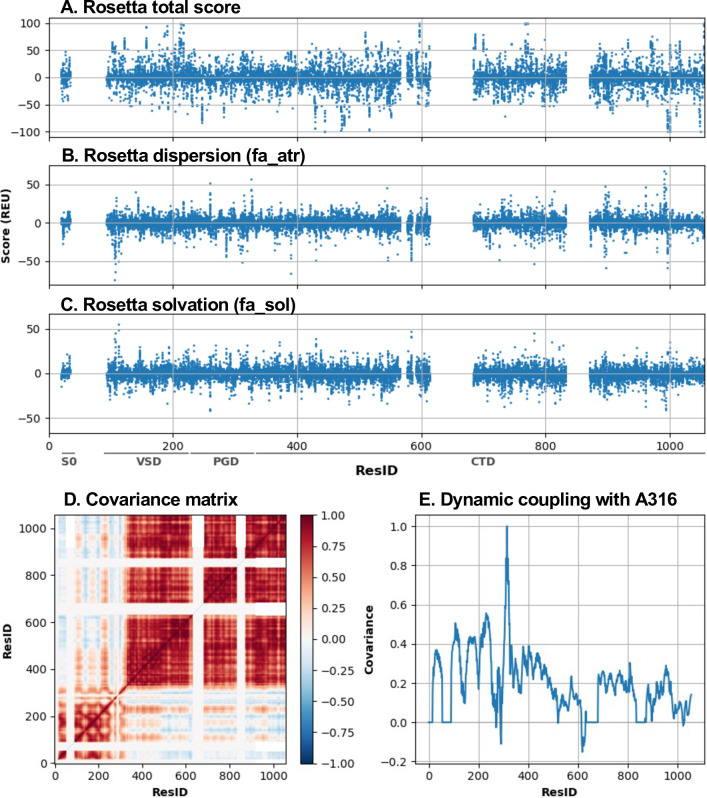
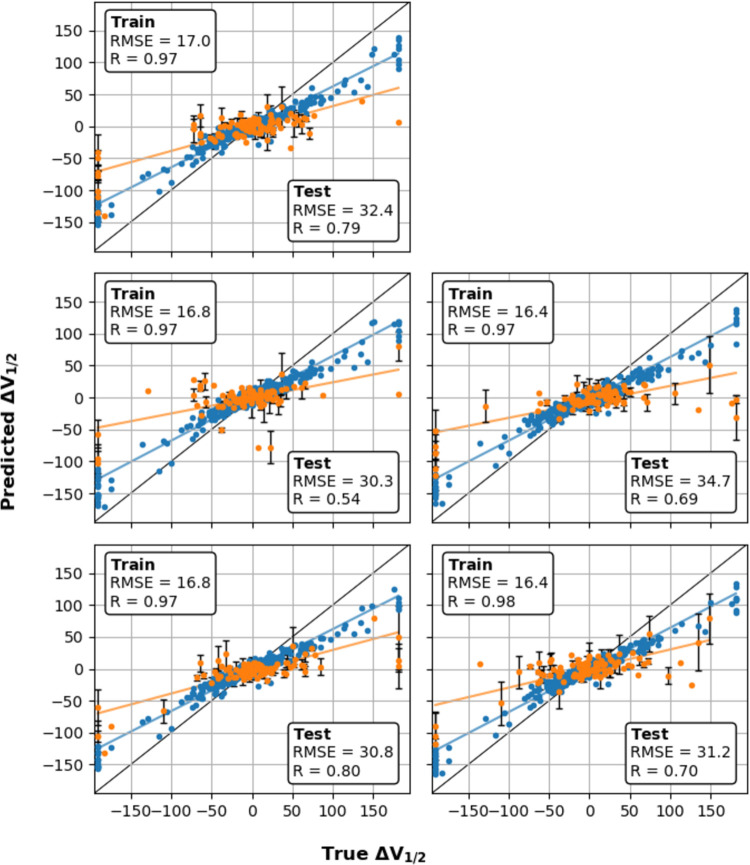
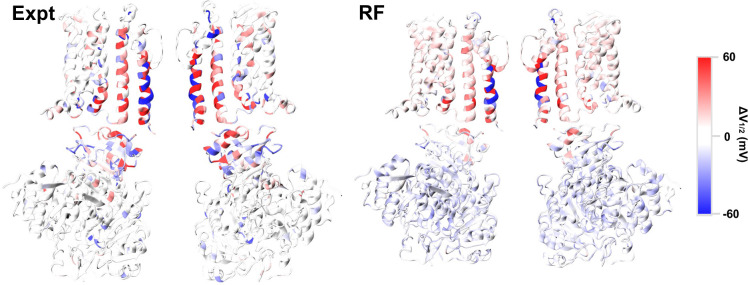
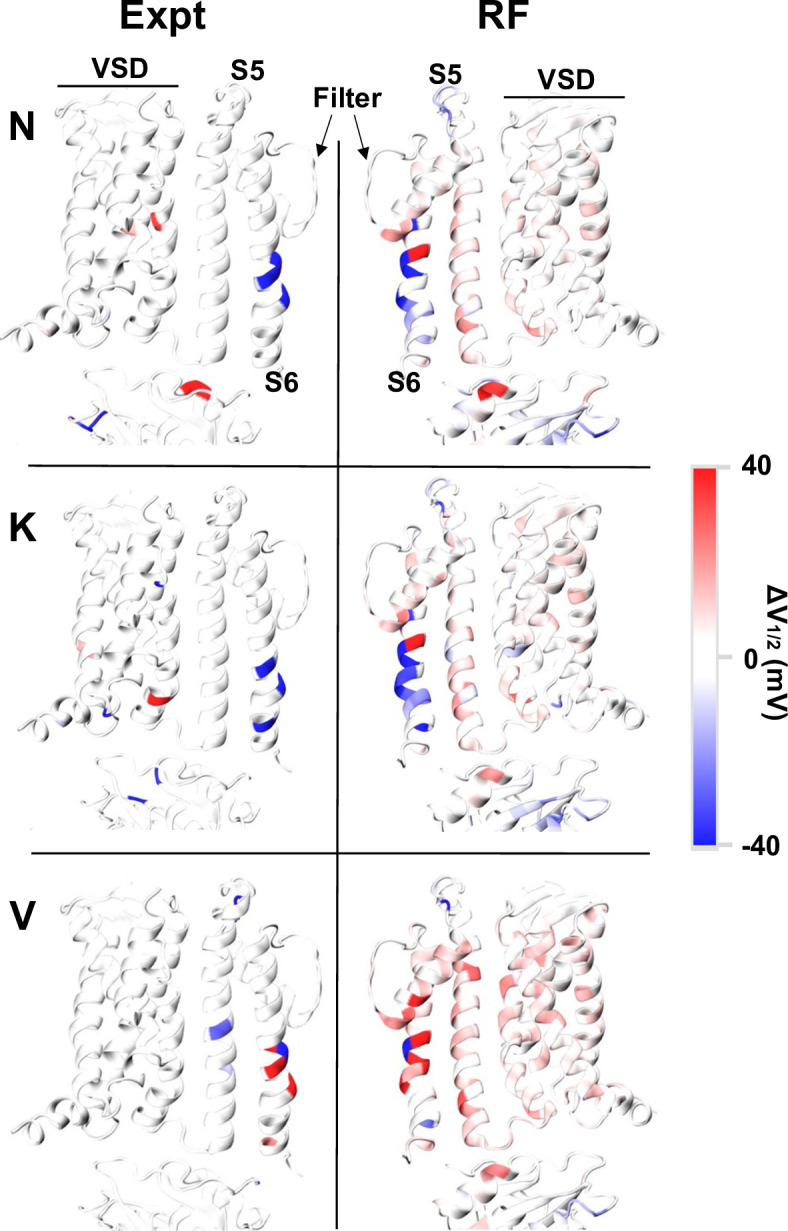
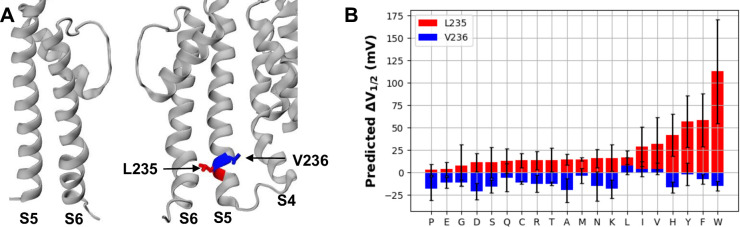
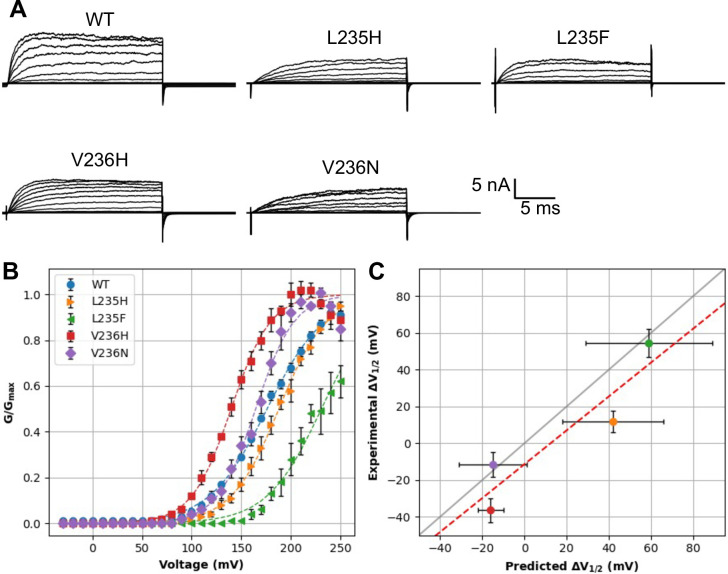
Machine learning has played transformative roles in numerous chemical and biophysical problems such as protein folding where large amount of data exists. Nonetheless, many important problems remain challenging for data-driven machine learning approaches due to the limitation of data scarcity. One approach to overcome data scarcity is to incorporate physical principles such as through molecular modeling and simulation. Here, we focus on the big potassium (BK) channels that play important roles in cardiovascular and neural systems. Many mutants of BK channel are associated with various neurological and cardiovascular diseases, but the molecular effects are unknown. The voltage gating properties of BK channels have been characterized for 473 site-specific mutations experimentally over the last three decades; yet, these functional data by themselves remain far too sparse to derive a predictive model of BK channel voltage gating. Using physics-based modeling, we quantify the energetic effects of all single mutations on both open and closed states of the channel. Together with dynamic properties derived from atomistic simulations, these physical descriptors allow the training of random forest models that could reproduce unseen experimentally measured shifts in gating voltage, ∆V1/2, with a RMSE ~ 32 mV and correlation coefficient of R ~ 0.7. Importantly, the model appears capable of uncovering nontrivial physical principles underlying the gating of the channel, including a central role of hydrophobic gating. The model was further evaluated using four novel mutations of L235 and V236 on the S5 helix, mutations of which are predicted to have opposing effects on V1/2 and suggest a key role of S5 in mediating voltage sensor-pore coupling. The measured ∆V1/2 agree quantitatively with prediction for all four mutations, with a high correlation of R = 0.92 and RMSE = 18 mV. Therefore, the model can capture nontrivial voltage gating properties in regions where few mutations are known. The success of predictive modeling of BK voltage gating demonstrates the potential of combining physics and statistical learning for overcoming data scarcity in nontrivial protein function prediction.
Copyright: © 2023 Nordquist et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Update of
-
Incorporating physics to overcome data scarcity in predictive modeling of protein function: a case study of BK channels.bioRxiv [Preprint]. 2023 Jun 26:2023.06.24.546384. doi: 10.1101/2023.06.24.546384. bioRxiv. 2023. Update in: PLoS Comput Biol. 2023 Sep 15;19(9):e1011460. doi: 10.1371/journal.pcbi.1011460. PMID: 37425916 Free PMC article. Updated. Preprint.
Similar articles
-
Incorporating physics to overcome data scarcity in predictive modeling of protein function: a case study of BK channels.bioRxiv [Preprint]. 2023 Jun 26:2023.06.24.546384. doi: 10.1101/2023.06.24.546384. bioRxiv. 2023. Update in: PLoS Comput Biol. 2023 Sep 15;19(9):e1011460. doi: 10.1371/journal.pcbi.1011460. PMID: 37425916 Free PMC article. Updated. Preprint.
-
A role for the S0 transmembrane segment in voltage-dependent gating of BK channels.J Gen Physiol. 2007 Mar;129(3):209-20. doi: 10.1085/jgp.200609662. Epub 2007 Feb 12. J Gen Physiol. 2007. PMID: 17296928 Free PMC article.
-
An S6 mutation in BK channels reveals beta1 subunit effects on intrinsic and voltage-dependent gating.J Gen Physiol. 2006 Dec;128(6):731-44. doi: 10.1085/jgp.200609596. J Gen Physiol. 2006. PMID: 17130522 Free PMC article.
-
Emerging issues of connexin channels: biophysics fills the gap.Q Rev Biophys. 2001 Aug;34(3):325-472. doi: 10.1017/s0033583501003705. Q Rev Biophys. 2001. PMID: 11838236 Review.
-
A BK (Slo1) channel journey from molecule to physiology.Channels (Austin). 2013 Nov-Dec;7(6):442-58. doi: 10.4161/chan.26242. Epub 2013 Sep 11. Channels (Austin). 2013. PMID: 24025517 Free PMC article. Review.
Cited by
-
Biophysics-based protein language models for protein engineering.bioRxiv [Preprint]. 2025 Jan 14:2024.03.15.585128. doi: 10.1101/2024.03.15.585128. bioRxiv. 2025. PMID: 38559182 Free PMC article. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
