

Tumor suppressor p53 mediates interleukin-6 expression to enable cancer cell evasion of genotoxic stress
- PMID: 37696858
- PMCID: PMC10495329
- DOI: 10.1038/s41420-023-01638-0
Tumor suppressor p53 mediates interleukin-6 expression to enable cancer cell evasion of genotoxic stress
Abstract
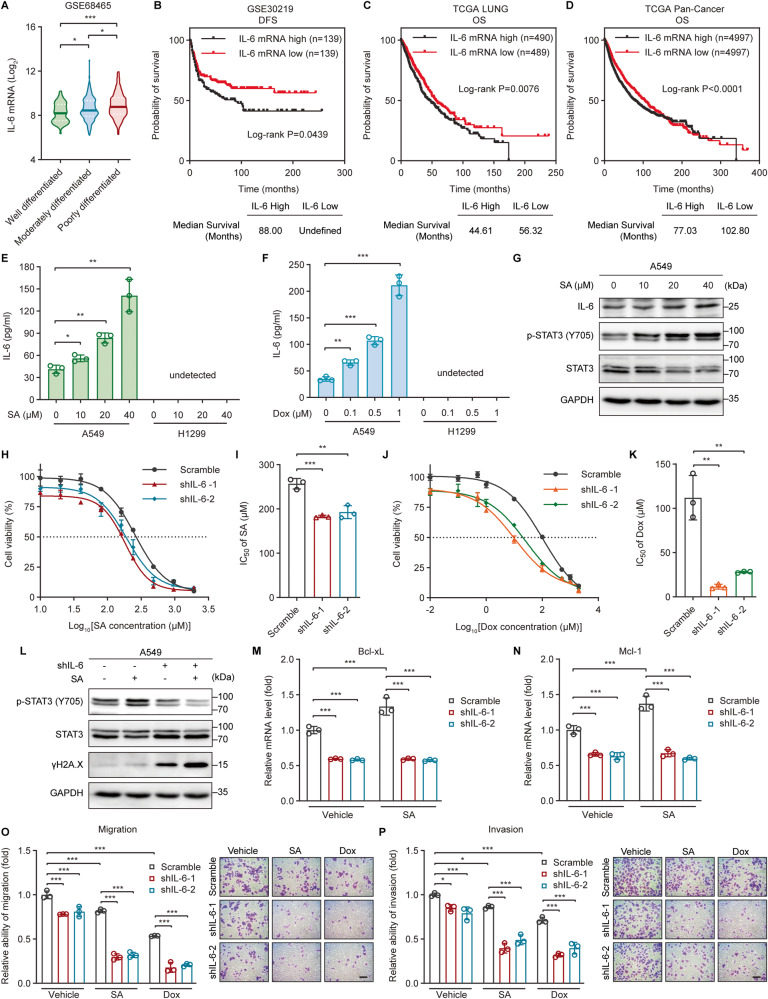
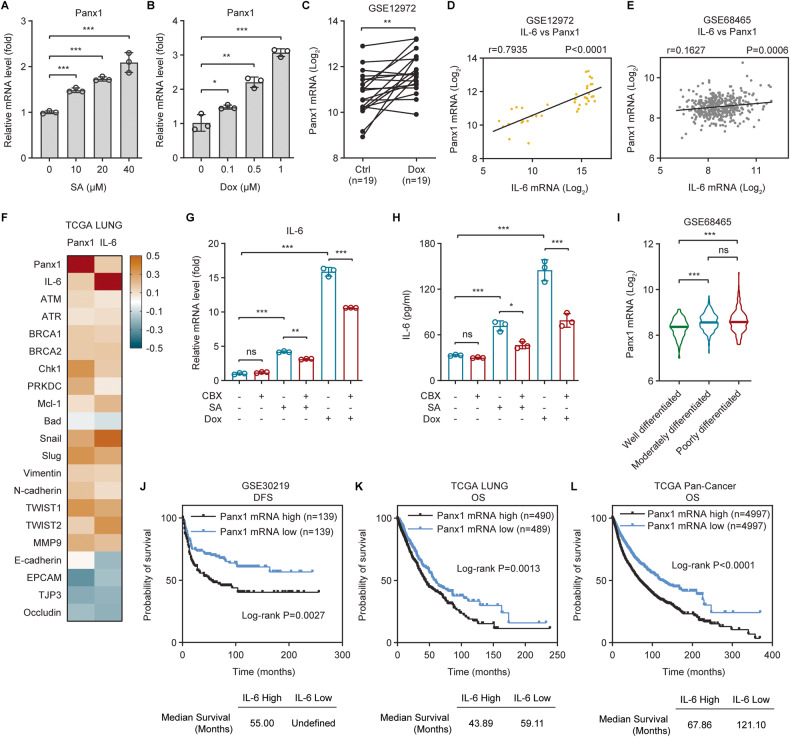
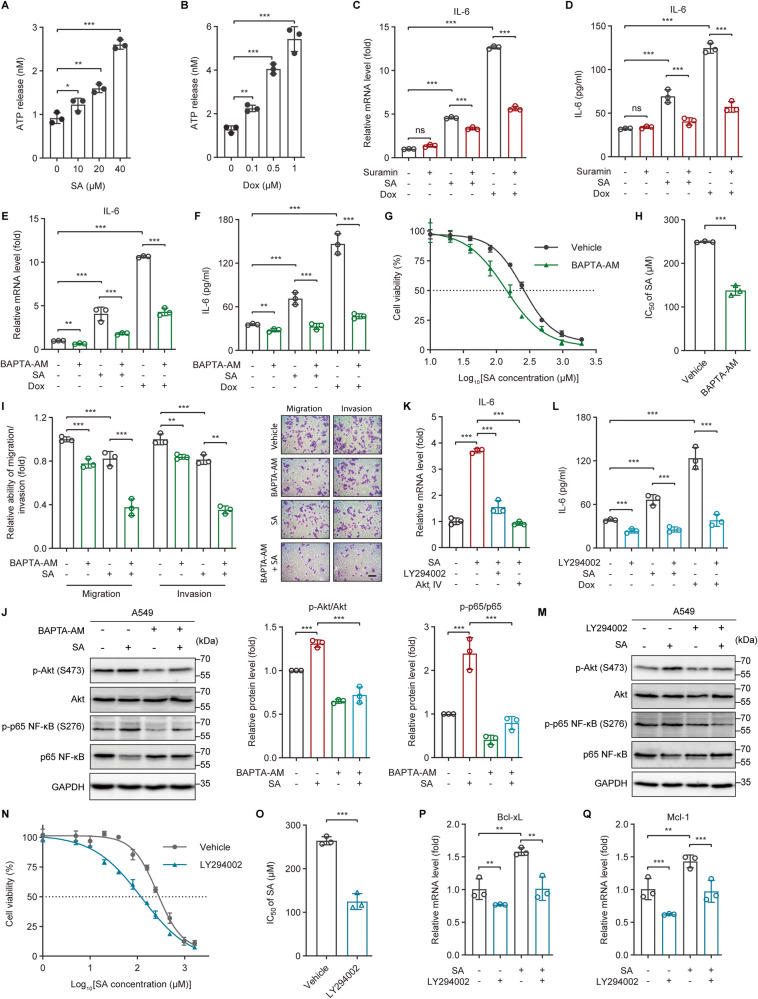
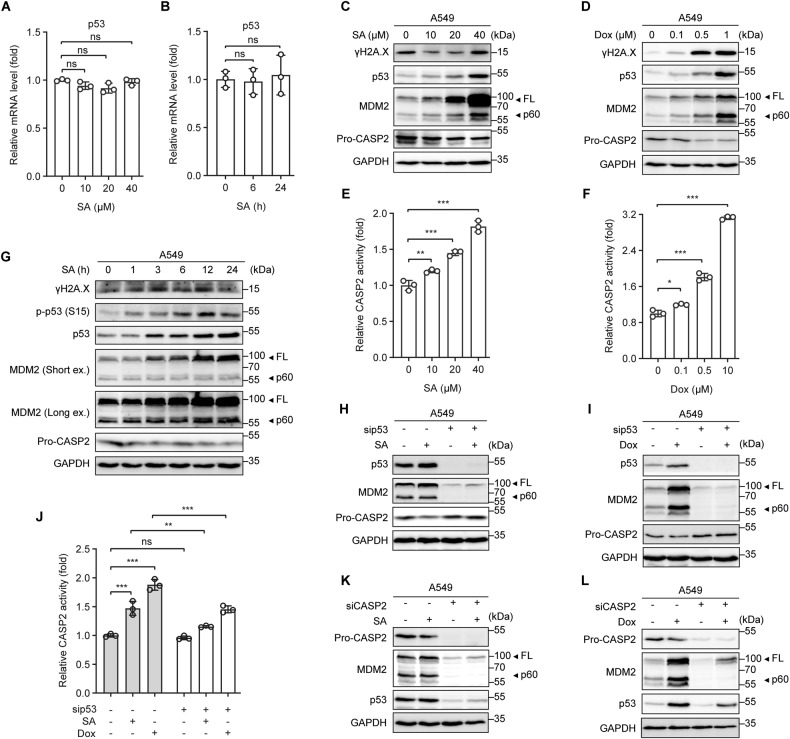
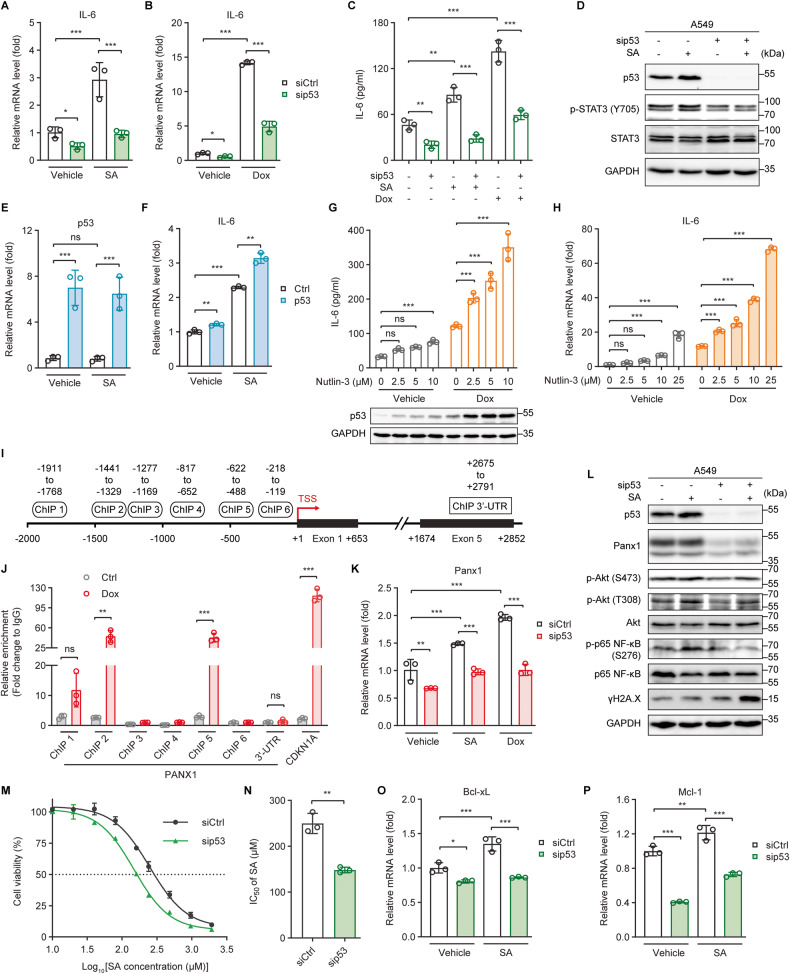
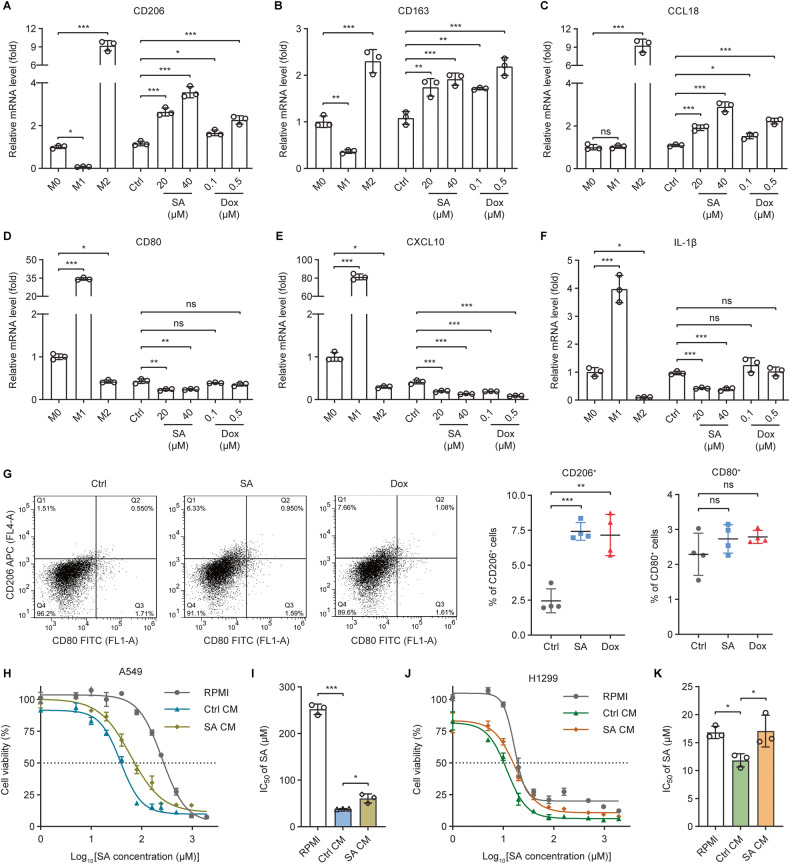
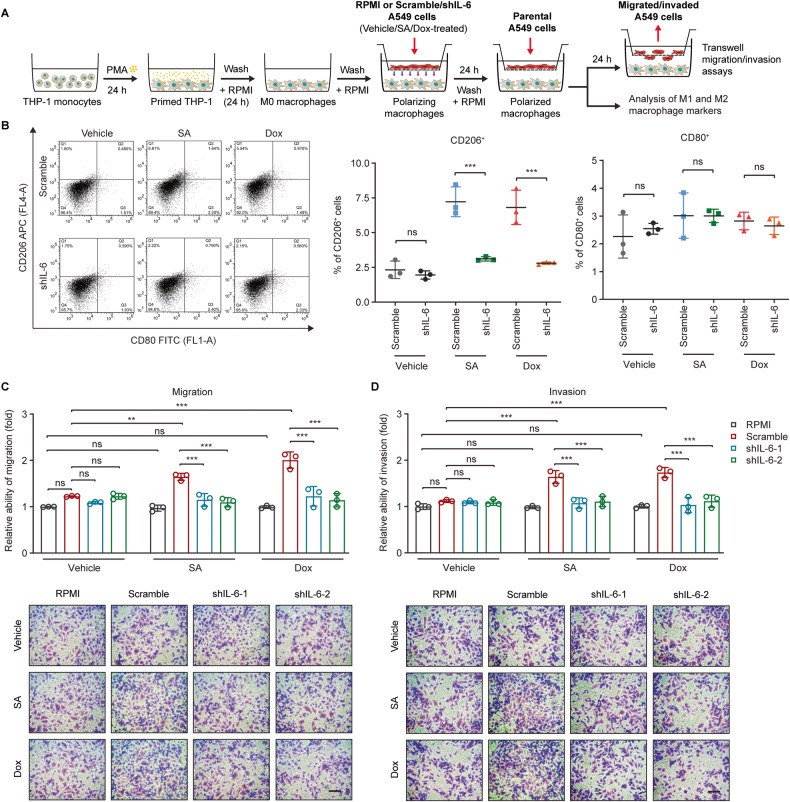
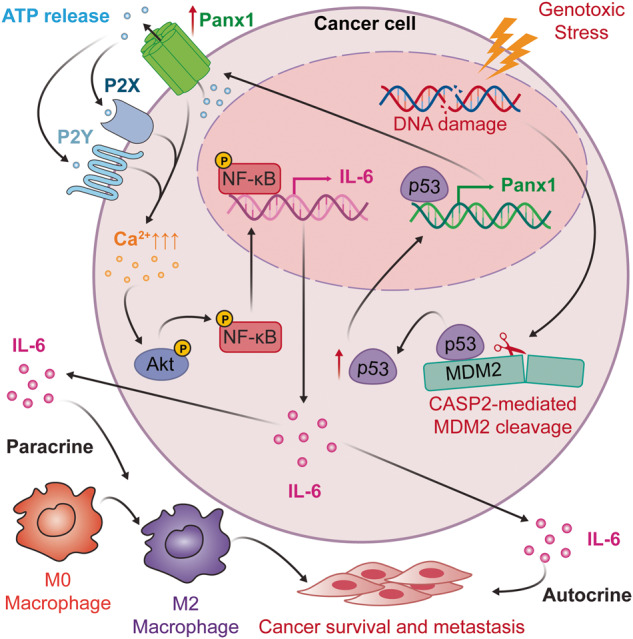
The tumor suppressor p53 primarily functions as a mediator of DNA damage-induced cell death, thereby contributing to the efficacy of genotoxic anticancer therapeutics. Here, we show, on the contrary, that cancer cells can employ genotoxic stress-induced p53 to acquire treatment resistance through the production of the pleiotropic cytokine interleukin (IL)-6. Mechanistically, DNA damage, either repairable or irreparable, activates p53 and stimulates Caspase-2-mediated cleavage of its negative regulator mouse double minute 2 (MDM2) creating a positive feedback loop that leads to elevated p53 protein accumulation. p53 transcriptionally controls the major adenosine triphosphate (ATP) release channel pannexin 1 (Panx1), which directs IL-6 induction via a mechanism dependent on the extracellular ATP-activated purinergic P2 receptors as well as their downstream intracellular calcium (iCa2+)/PI3K/Akt/NF-ĸB signaling pathway. Thus, p53 silencing impairs Panx1 and IL-6 expression and renders cancer cells sensitive to genotoxic stress. Moreover, we confirm that IL-6 hampers the effectiveness of genotoxic anticancer agents by mitigating DNA damage, driving the expression of anti-apoptotic Bcl-2 family genes, and maintaining the migratory and invasive properties of cancer cells. Analysis of patient survival and relevant factors in lung cancer and pan-cancer cohorts supports the prognostic and clinical values of Panx1 and IL-6. Notably, IL-6 secreted by cancer cells during genotoxic treatments promotes the polarization of monocytic THP-1-derived macrophages into an alternative (M2-like) phenotype that exhibits impaired anti-survival activities but enhanced pro-metastatic effects on cancer cells as compared to nonpolarized macrophages. Our study reveals the precise mechanism for genotoxic-induced IL-6 and suggests that targeting p53-mediated IL-6 may improve the responsiveness of cancer cells to genotoxic anticancer therapy.
© 2023. Cell Death Differentiation Association (ADMC).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage.J Immunol. 2008 Jun 1;180(11):7147-57. doi: 10.4049/jimmunol.180.11.7147. J Immunol. 2008. PMID: 18490713
-
Chemotherapeutic drugs induce ATP release via caspase-gated pannexin-1 channels and a caspase/pannexin-1-independent mechanism.J Biol Chem. 2014 Sep 26;289(39):27246-27263. doi: 10.1074/jbc.M114.590240. Epub 2014 Aug 11. J Biol Chem. 2014. PMID: 25112874 Free PMC article.
-
Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop.Mol Cell. 2011 Jul 8;43(1):57-71. doi: 10.1016/j.molcel.2011.06.012. Mol Cell. 2011. PMID: 21726810 Free PMC article.
-
A systematic review of p53 regulation of oxidative stress in skeletal muscle.Redox Rep. 2018 Dec;23(1):100-117. doi: 10.1080/13510002.2017.1416773. Epub 2018 Jan 3. Redox Rep. 2018. PMID: 29298131 Free PMC article. Review.
-
A new twist in the feedback loop: stress-activated MDM2 destabilization is required for p53 activation.Cell Cycle. 2005 Mar;4(3):411-7. doi: 10.4161/cc.4.3.1522. Epub 2005 Mar 2. Cell Cycle. 2005. PMID: 15684615 Review.
Cited by
-
Inverse correlation between TP53 gene status and PD-L1 protein levels in a melanoma cell model depends on an IRF1/SOX10 regulatory axis.Cell Mol Biol Lett. 2024 Sep 5;29(1):117. doi: 10.1186/s11658-024-00637-y. Cell Mol Biol Lett. 2024. PMID: 39237877 Free PMC article.
-
Synthesis, characterization and biological research of novel 2-(quinoline-4-carbonyl)hydrazide-acrylamide hybrids as potential anticancer agents on MCF-7 breast carcinoma cells by targeting EGFR-TK.RSC Adv. 2024 Jul 26;14(32):23495-23504. doi: 10.1039/d4ra03963g. eCollection 2024 Jul 19. RSC Adv. 2024. PMID: 39071480 Free PMC article.
-
p38 Molecular Targeting for Next-Generation Multiple Myeloma Therapy.Cancers (Basel). 2024 Jan 6;16(2):256. doi: 10.3390/cancers16020256. Cancers (Basel). 2024. PMID: 38254747 Free PMC article. Review.
-
Bioinformatics analysis and validation of CSRNP1 as a key prognostic gene in non-small cell lung cancer.Heliyon. 2024 Mar 19;10(7):e28412. doi: 10.1016/j.heliyon.2024.e28412. eCollection 2024 Apr 15. Heliyon. 2024. PMID: 38560128 Free PMC article.
References
-
- Seddon B, Strauss SJ, Whelan J, Leahy M, Woll PJ, Cowie F, et al. Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): a randomised controlled phase 3 trial. Lancet Oncol. 2017;18:1397–410. doi: 10.1016/S1470-2045(17)30622-8. - DOI - PMC - PubMed
-
- Gridelli C, Morabito A, Cavanna L, Luciani A, Maione P, Bonanno L, et al. Cisplatin-based first-line treatment of elderly patients with advanced non–small-cell lung cancer: joint analysis of MILES-3 and MILES-4 phase III trials. J Clin Oncol. 2018;36:2585–92. doi: 10.1200/JCO.2017.76.8390. - DOI - PubMed
Grants and funding
- 109D0042V1/National Tsing Hua University (NTHU)
- 109D0042V1/National Tsing Hua University (NTHU)
- 109D0042V1/National Tsing Hua University (NTHU)
- 108-2311-B-007-008/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
- 108-2311-B-007-008/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
