

The virome of the invasive Asian bush mosquito Aedes japonicus in Europe
- PMID: 37636319
- PMCID: PMC10460169
- DOI: 10.1093/ve/vead041
The virome of the invasive Asian bush mosquito Aedes japonicus in Europe
Abstract
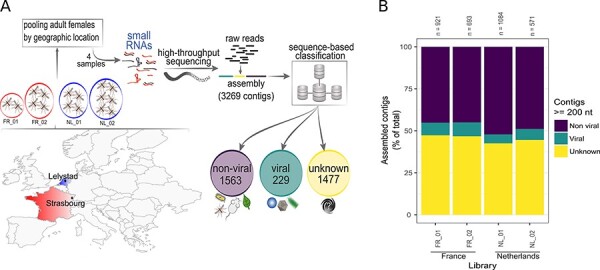
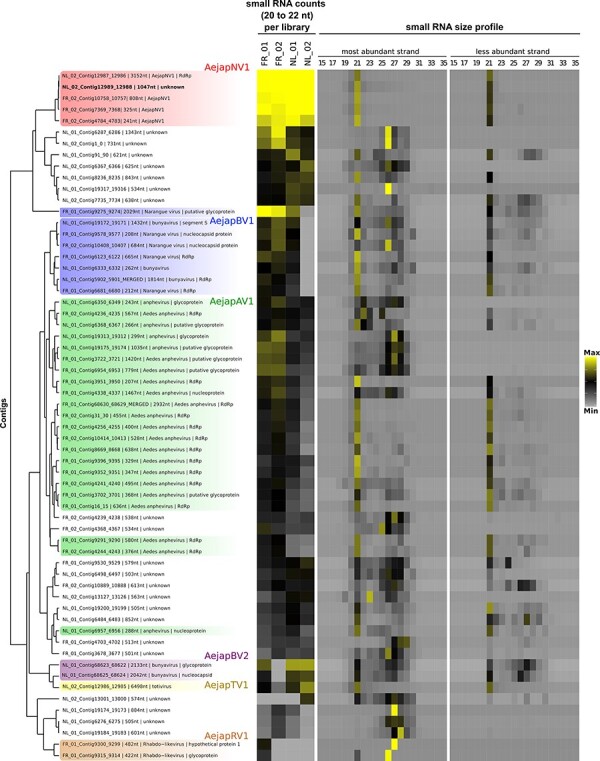
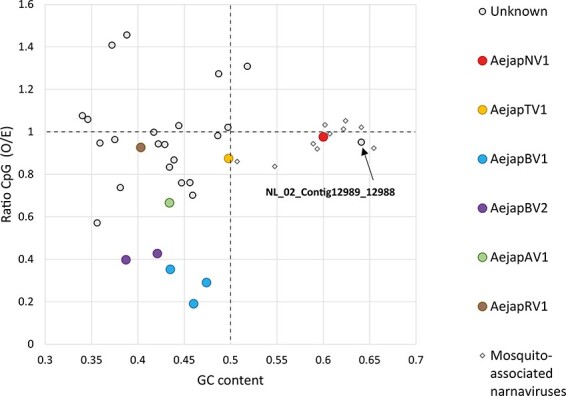
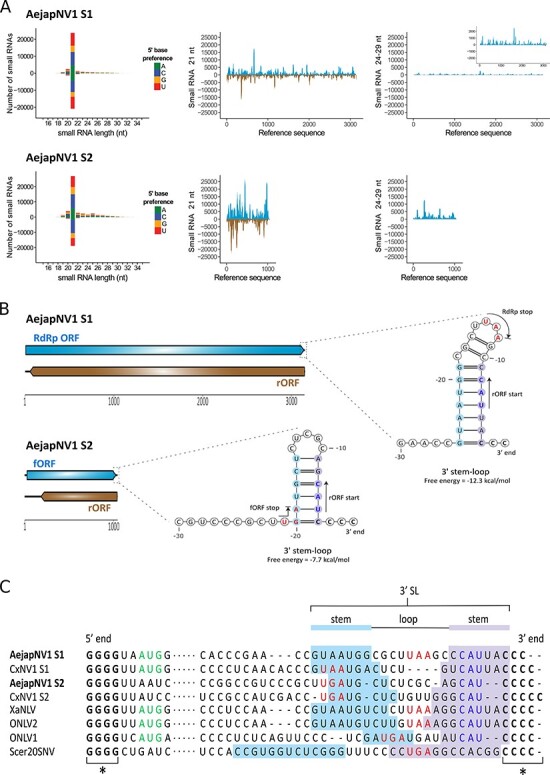
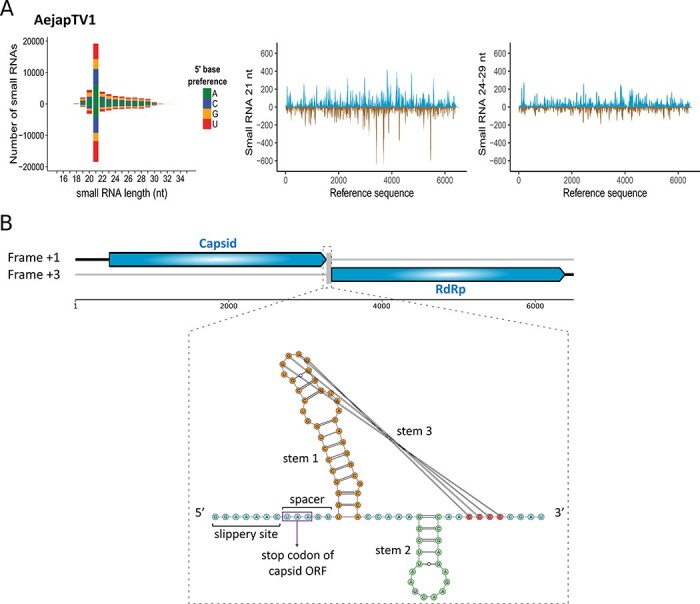
The Asian bush mosquito Aedes japonicus is rapidly invading North America and Europe. Due to its potential to transmit multiple pathogenic arthropod-borne (arbo)viruses including Zika virus, West Nile virus, and chikungunya virus, it is important to understand the biology of this vector mosquito in more detail. In addition to arboviruses, mosquitoes can also carry insect-specific viruses that are receiving increasing attention due to their potential effects on host physiology and arbovirus transmission. In this study, we characterized the collection of viruses, referred to as the virome, circulating in Ae. japonicus populations in the Netherlands and France. Applying a small RNA-based metagenomic approach to Ae. japonicus, we uncovered a distinct group of viruses present in samples from both the Netherlands and France. These included one known virus, Ae. japonicus narnavirus 1 (AejapNV1), and three new virus species that we named Ae. japonicus totivirus 1 (AejapTV1), Ae. japonicus anphevirus 1 (AejapAV1) and Ae. japonicus bunyavirus 1 (AejapBV1). We also discovered sequences that were presumably derived from two additional novel viruses: Ae. japonicus bunyavirus 2 (AejapBV2) and Ae. japonicus rhabdovirus 1 (AejapRV1). All six viruses induced strong RNA interference responses, including the production of twenty-one nucleotide-sized small interfering RNAs, a signature of active replication in the host. Notably, AejapBV1 and AejapBV2 belong to different viral families; however, no RNA-dependent RNA polymerase sequence has been found for AejapBV2. Intriguingly, our small RNA-based approach identified an ∼1-kb long ambigrammatic RNA that is associated with AejapNV1 as a secondary segment but showed no similarity to any sequence in public databases. We confirmed the presence of AejapNV1 primary and secondary segments, AejapTV1, AejapAV1, and AejapBV1 by reverse transcriptase polymerase chain reaction (PCR) in wild-caught Ae. japonicus mosquitoes. AejapNV1 and AejapTV1 were found at high prevalence (87-100 per cent) in adult females, adult males, and larvae. Using a small RNA-based, sequence-independent metagenomic strategy, we uncovered a conserved and prevalent virome among Ae. japonicus mosquito populations. The high prevalence of AejapNV1 and AejapTV1 across all tested mosquito life stages suggests that these viruses are intimately associated with Ae. japonicus.
Keywords: Aedes japonicus; RNA interference; anphevirus; bunyavirus; metagenomics; mosquito; narnavirus; rhabdovirus; totivirus; virome.
© The Author(s) 2023. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures





Similar articles
-
The invasive Asian bush mosquito Aedes japonicus found in the Netherlands can experimentally transmit Zika virus and Usutu virus.PLoS Negl Trop Dis. 2020 Apr 13;14(4):e0008217. doi: 10.1371/journal.pntd.0008217. eCollection 2020 Apr. PLoS Negl Trop Dis. 2020. PMID: 32282830 Free PMC article.
-
Introduction of invasive mosquito species into Europe and prospects for arbovirus transmission and vector control in an era of globalization.Infect Dis Poverty. 2023 Nov 30;12(1):109. doi: 10.1186/s40249-023-01167-z. Infect Dis Poverty. 2023. PMID: 38037192 Free PMC article.
-
Spatiotemporal distribution, abundance, and host interactions of two invasive vectors of arboviruses, Aedes albopictus and Aedes japonicus, in Pennsylvania, USA.Parasit Vectors. 2022 Jan 24;15(1):36. doi: 10.1186/s13071-022-05151-8. Parasit Vectors. 2022. PMID: 35073977 Free PMC article.
-
Aedes aegypti and Ae. albopictus microbiome/virome: new strategies for controlling arboviral transmission?Parasit Vectors. 2022 Aug 9;15(1):287. doi: 10.1186/s13071-022-05401-9. Parasit Vectors. 2022. PMID: 35945559 Free PMC article. Review.
-
The potential invasion into North America and Europe by non-native mosquito, Aedes koreicus (Diptera: Culicidae).J Med Entomol. 2023 Nov 14;60(6):1305-1313. doi: 10.1093/jme/tjad116. J Med Entomol. 2023. PMID: 37651733 Review.
Cited by
-
Transcriptome Analysis Reveals a Diverse Range of Novel Viruses in Australian Sugarcane Soldier Fly (Inopus flavus) Larvae.Viruses. 2024 Mar 27;16(4):516. doi: 10.3390/v16040516. Viruses. 2024. PMID: 38675859 Free PMC article.
-
Editorial: Using virus specific-signatures during infection to characterize host-pathogen interactions.Front Genet. 2023 Sep 22;14:1290714. doi: 10.3389/fgene.2023.1290714. eCollection 2023. Front Genet. 2023. PMID: 37811151 Free PMC article. No abstract available.
-
The potential role of the Asian bush mosquito Aedes japonicus as spillover vector for West Nile virus in the Netherlands.Parasit Vectors. 2024 Jun 17;17(1):262. doi: 10.1186/s13071-024-06279-5. Parasit Vectors. 2024. PMID: 38886805 Free PMC article.
-
Lack of abundant core virome in Culex mosquitoes from a temperate climate region despite a mosquito species-specific virome.mSystems. 2024 Jun 18;9(6):e0001224. doi: 10.1128/msystems.00012-24. Epub 2024 May 14. mSystems. 2024. PMID: 38742876 Free PMC article.
-
A parasite odyssey: An RNA virus concealed in Toxoplasma gondii.Virus Evol. 2024 May 11;10(1):veae040. doi: 10.1093/ve/veae040. eCollection 2024. Virus Evol. 2024. PMID: 38817668 Free PMC article.
References
LinkOut - more resources
Full Text Sources
