

ChimeraTE: a pipeline to detect chimeric transcripts derived from genes and transposable elements
- PMID: 37615575
- PMCID: PMC10570057
- DOI: 10.1093/nar/gkad671
ChimeraTE: a pipeline to detect chimeric transcripts derived from genes and transposable elements
Abstract
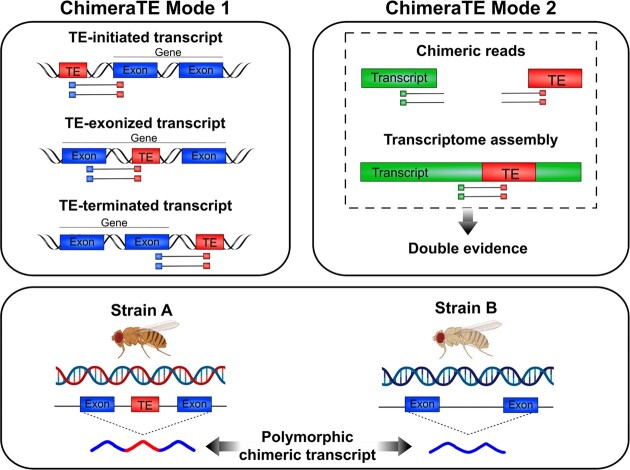
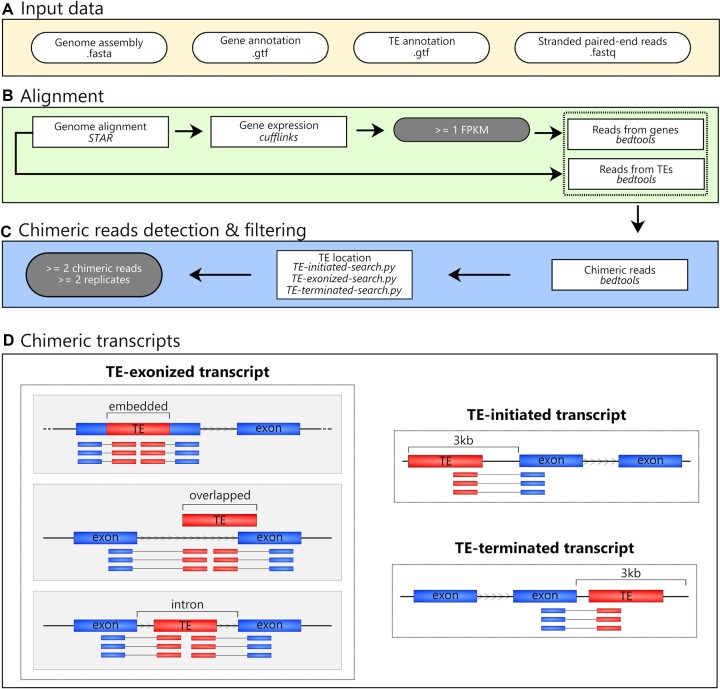
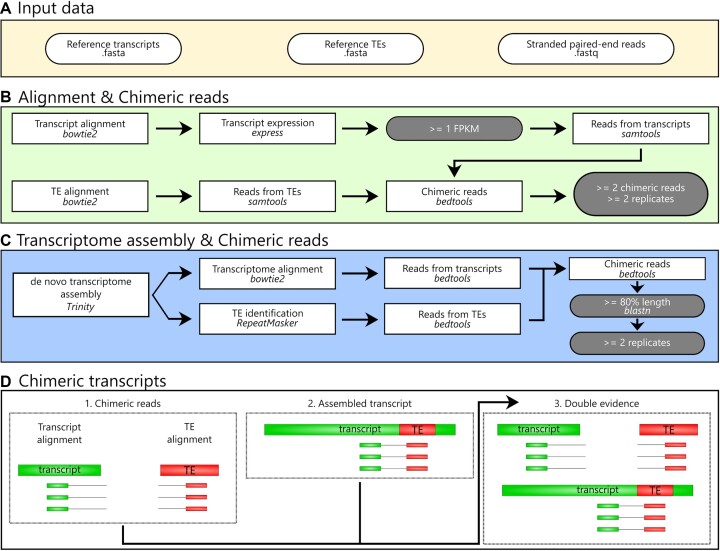
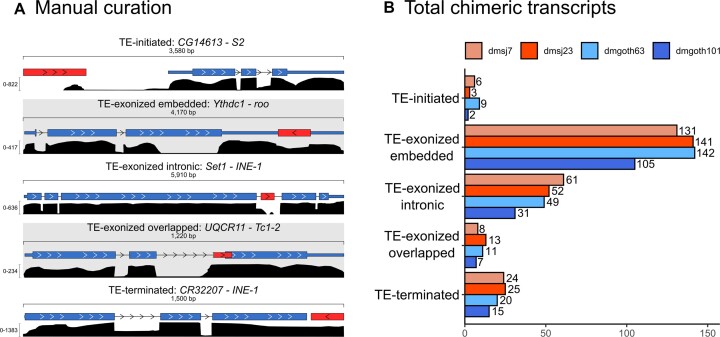
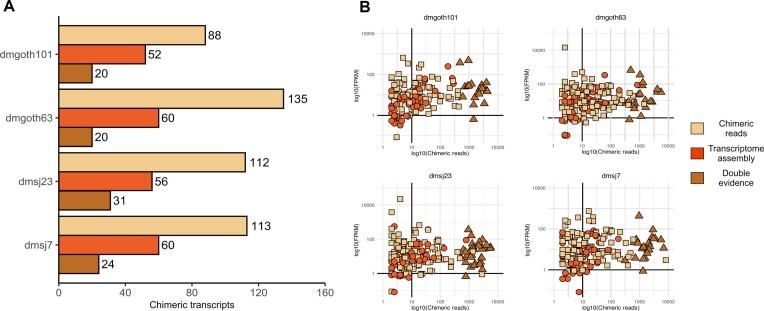
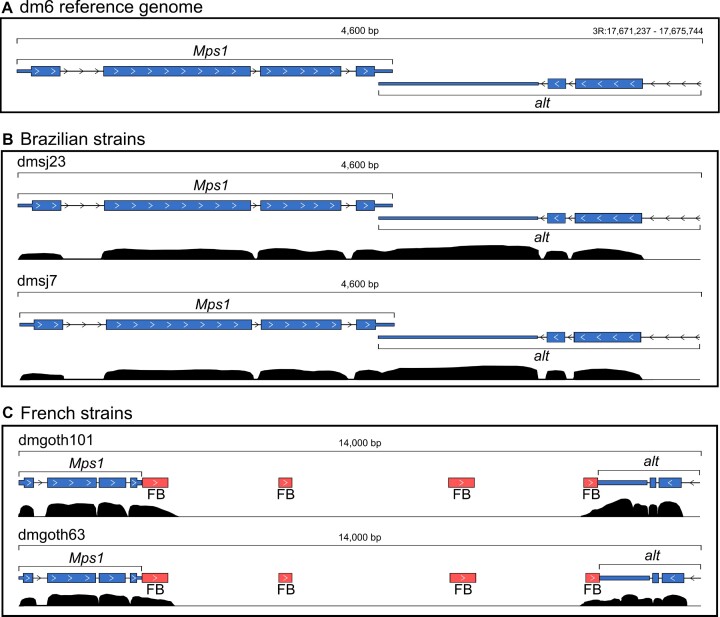
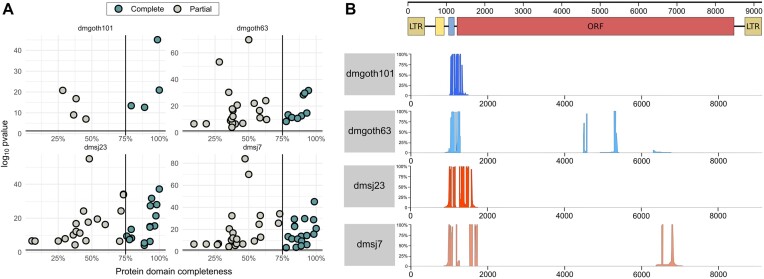
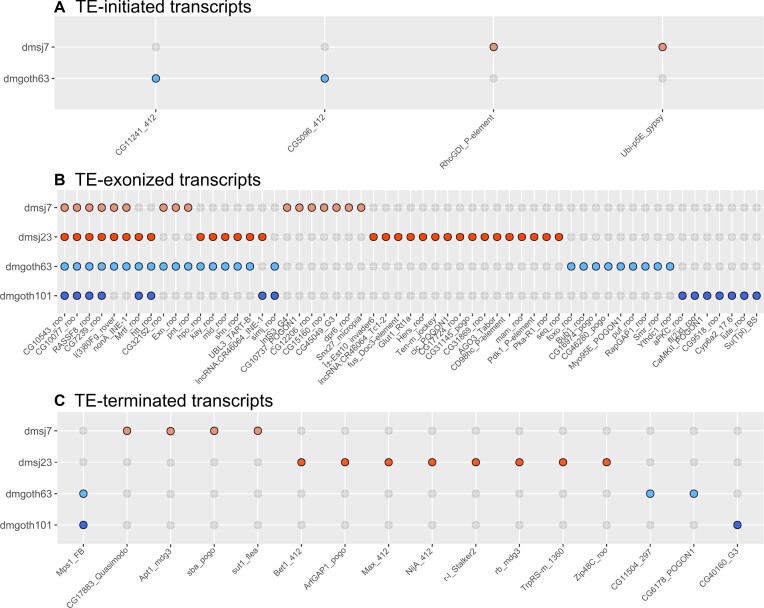
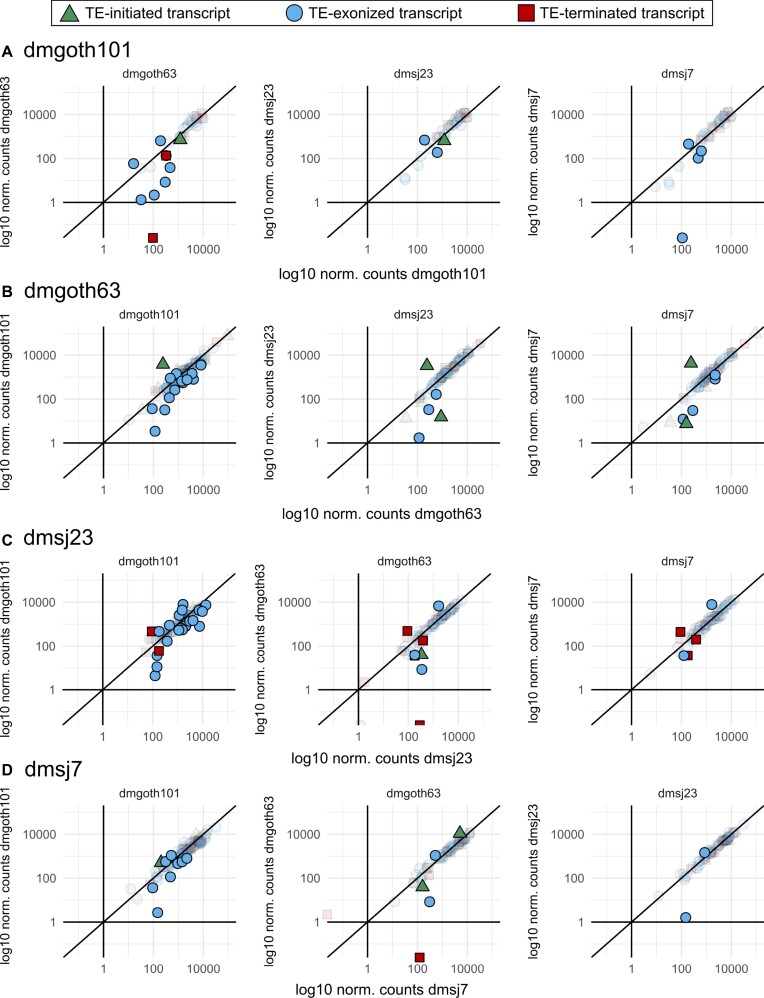
Transposable elements (TEs) produce structural variants and are considered an important source of genetic diversity. Notably, TE-gene fusion transcripts, i.e. chimeric transcripts, have been associated with adaptation in several species. However, the identification of these chimeras remains hindered due to the lack of detection tools at a transcriptome-wide scale, and to the reliance on a reference genome, even though different individuals/cells/strains have different TE insertions. Therefore, we developed ChimeraTE, a pipeline that uses paired-end RNA-seq reads to identify chimeric transcripts through two different modes. Mode 1 is the reference-guided approach that employs canonical genome alignment, and Mode 2 identifies chimeras derived from fixed or insertionally polymorphic TEs without any reference genome. We have validated both modes using RNA-seq data from four Drosophila melanogaster wild-type strains. We found ∼1.12% of all genes generating chimeric transcripts, most of them from TE-exonized sequences. Approximately ∼23% of all detected chimeras were absent from the reference genome, indicating that TEs belonging to chimeric transcripts may be recent, polymorphic insertions. ChimeraTE is the first pipeline able to automatically uncover chimeric transcripts without a reference genome, consisting of two running Modes that can be used as a tool to investigate the contribution of TEs to transcriptome plasticity.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

Similar articles
-
Transposons contribute to the functional diversification of the head, gut, and ovary transcriptomes across Drosophila natural strains.Genome Res. 2023 Sep;33(9):1541-1553. doi: 10.1101/gr.277565.122. Epub 2023 Oct 4. Genome Res. 2023. PMID: 37793782 Free PMC article.
-
Paucity of chimeric gene-transposable element transcripts in the Drosophila melanogaster genome.BMC Biol. 2005 Nov 12;3:24. doi: 10.1186/1741-7007-3-24. BMC Biol. 2005. PMID: 16283942 Free PMC article.
-
Analysis of RNA-Seq Data Using TEtranscripts.Methods Mol Biol. 2018;1751:153-167. doi: 10.1007/978-1-4939-7710-9_11. Methods Mol Biol. 2018. PMID: 29508296
-
Measuring and interpreting transposable element expression.Nat Rev Genet. 2020 Dec;21(12):721-736. doi: 10.1038/s41576-020-0251-y. Epub 2020 Jun 23. Nat Rev Genet. 2020. PMID: 32576954 Review.
-
Population genomics of transposable elements in Drosophila.Annu Rev Genet. 2014;48:561-81. doi: 10.1146/annurev-genet-120213-092359. Epub 2014 Oct 1. Annu Rev Genet. 2014. PMID: 25292358 Review.
Cited by
-
Technology to the rescue: how to uncover the role of transposable elements in preimplantation development.Biochem Soc Trans. 2024 Jun 26;52(3):1349-1362. doi: 10.1042/BST20231262. Biochem Soc Trans. 2024. PMID: 38752836 Free PMC article. Review.
-
Transposons contribute to the functional diversification of the head, gut, and ovary transcriptomes across Drosophila natural strains.Genome Res. 2023 Sep;33(9):1541-1553. doi: 10.1101/gr.277565.122. Epub 2023 Oct 4. Genome Res. 2023. PMID: 37793782 Free PMC article.
-
Transposable Element (TE) insertion predictions from RNAseq inputs and TE impact on RNA splicing and gene expression in Drosophila brain transcriptomes.Mob DNA. 2024 Oct 9;15(1):20. doi: 10.1186/s13100-024-00330-z. Mob DNA. 2024. PMID: 39385293 Free PMC article.
-
Investigation of chimeric transcripts derived from LINE-1 and Alu retrotransposons in cerebellar tissues of individuals with autism spectrum disorder (ASD).Sci Rep. 2024 Sep 19;14(1):21889. doi: 10.1038/s41598-024-72334-x. Sci Rep. 2024. PMID: 39300110 Free PMC article.
References
-
- International Human Genome Sequencing Consortium, Whitehead Institute for Biomedical Research, Center for Genome Research Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K.et al. .. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. - PubMed
-
- Danilevskaya O.N., Arkhipova I.R., Pardue M.L., Traverse K.L.. Promoting in tandem: the promoter for telomere transposon HeT-A and implications for the evolution of retroviral ltrs. Cell. 1997; 88:647–655. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
