

The HSV-1 ICP22 protein selectively impairs histone repositioning upon Pol II transcription downstream of genes
- PMID: 37524699
- PMCID: PMC10390501
- DOI: 10.1038/s41467-023-40217-w
The HSV-1 ICP22 protein selectively impairs histone repositioning upon Pol II transcription downstream of genes
Abstract
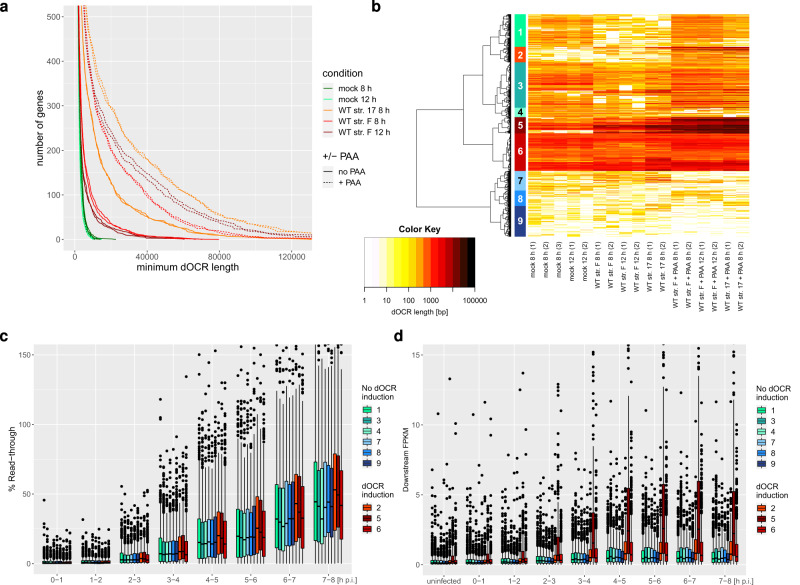
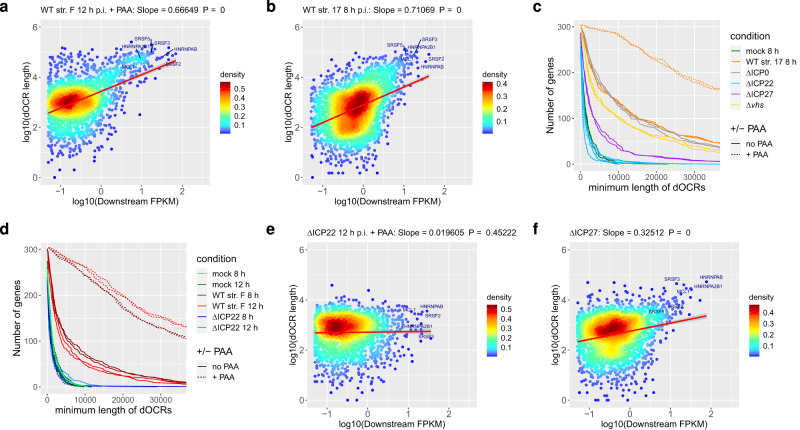
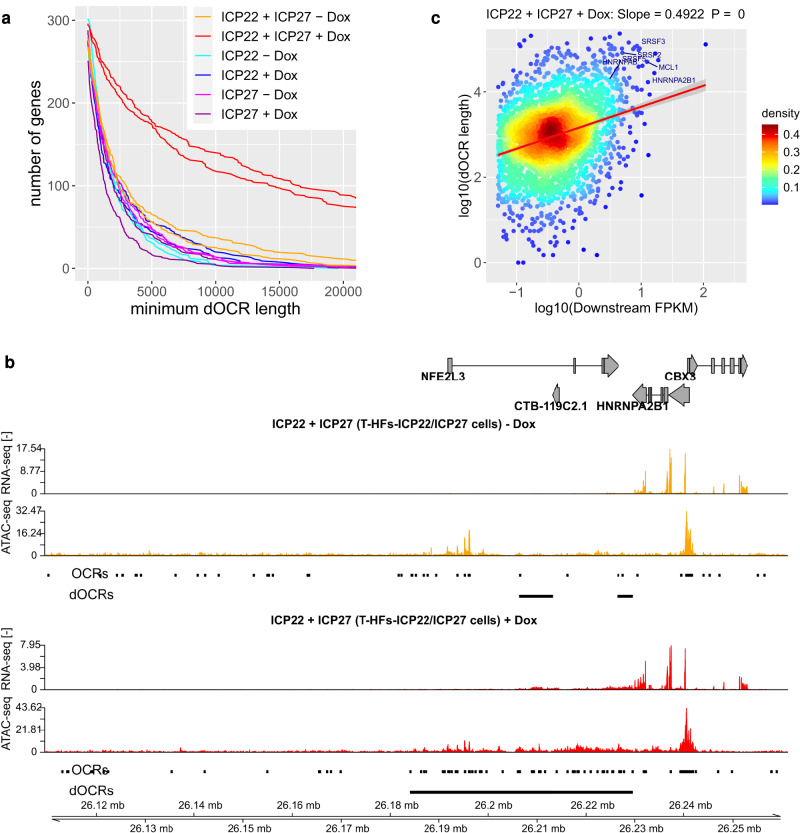
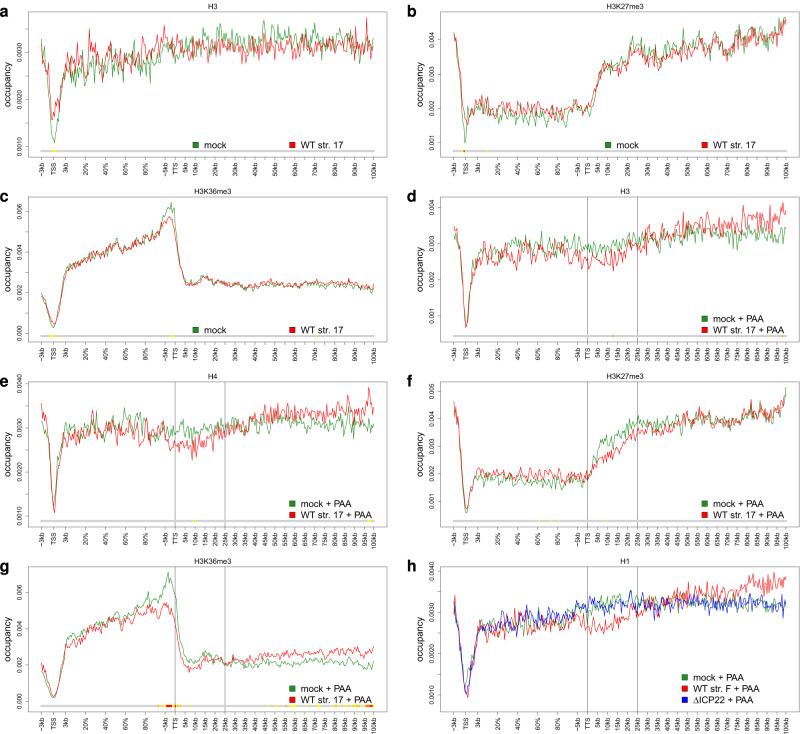
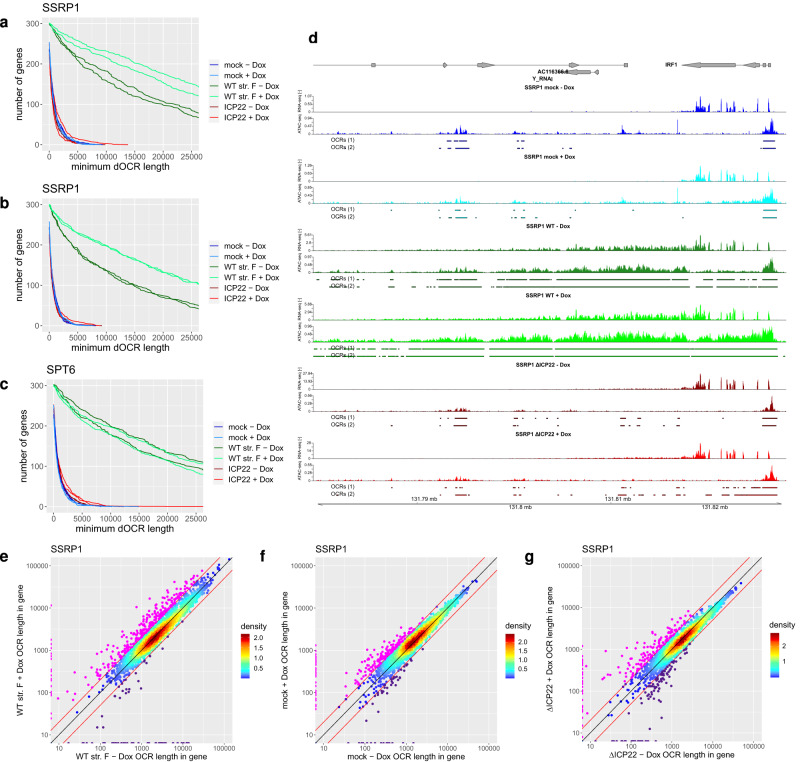
Herpes simplex virus 1 (HSV-1) infection and stress responses disrupt transcription termination by RNA Polymerase II (Pol II). In HSV-1 infection, but not upon salt or heat stress, this is accompanied by a dramatic increase in chromatin accessibility downstream of genes. Here, we show that the HSV-1 immediate-early protein ICP22 is both necessary and sufficient to induce downstream open chromatin regions (dOCRs) when transcription termination is disrupted by the viral ICP27 protein. This is accompanied by a marked ICP22-dependent loss of histones downstream of affected genes consistent with impaired histone repositioning in the wake of Pol II. Efficient knock-down of the ICP22-interacting histone chaperone FACT is not sufficient to induce dOCRs in ΔICP22 infection but increases dOCR induction in wild-type HSV-1 infection. Interestingly, this is accompanied by a marked increase in chromatin accessibility within gene bodies. We propose a model in which allosteric changes in Pol II composition downstream of genes and ICP22-mediated interference with FACT activity explain the differential impairment of histone repositioning downstream of genes in the wake of Pol II in HSV-1 infection.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
A Herpesviral Immediate Early Protein Promotes Transcription Elongation of Viral Transcripts.mBio. 2017 Jun 13;8(3):e00745-17. doi: 10.1128/mBio.00745-17. mBio. 2017. PMID: 28611249 Free PMC article.
-
Herpes simplex virus 1 inhibits phosphorylation of RNA polymerase II CTD serine-7.J Virol. 2024 Oct 22;98(10):e0117824. doi: 10.1128/jvi.01178-24. Epub 2024 Sep 24. J Virol. 2024. PMID: 39316591 Free PMC article.
-
ICP22 of Herpes Simplex Virus 1 Decreases RNA Polymerase Processivity.J Virol. 2022 Mar 9;96(5):e0219121. doi: 10.1128/jvi.02191-21. Epub 2022 Jan 12. J Virol. 2022. PMID: 35019725 Free PMC article.
-
A Review of the Multipronged Attack of Herpes Simplex Virus 1 on the Host Transcriptional Machinery.Viruses. 2021 Sep 14;13(9):1836. doi: 10.3390/v13091836. Viruses. 2021. PMID: 34578417 Free PMC article. Review.
-
The herpes simplex virus type 1 infected cell protein 22.Virol Sin. 2010 Feb;25(1):1-7. doi: 10.1007/s12250-010-3080-x. Epub 2010 Feb 12. Virol Sin. 2010. PMID: 20960278 Free PMC article. Review.
Cited by
-
A Revision of Herpes Simplex Virus Type 1 Transcription: First, Repress; Then, Express.Microorganisms. 2024 Jan 26;12(2):262. doi: 10.3390/microorganisms12020262. Microorganisms. 2024. PMID: 38399666 Free PMC article. Review.
-
Impact of the interaction between herpes simplex virus 1 ICP22 and FACT on viral gene expression and pathogenesis.J Virol. 2024 Aug 20;98(8):e0073724. doi: 10.1128/jvi.00737-24. Epub 2024 Jul 17. J Virol. 2024. PMID: 39016551 Free PMC article.
-
HSV-1 Infection Induces a Downstream Shift of Promoter-Proximal Pausing for Host Genes.J Virol. 2023 May 31;97(5):e0038123. doi: 10.1128/jvi.00381-23. Epub 2023 Apr 24. J Virol. 2023. PMID: 37093003 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
