

SOURSOP: A Python Package for the Analysis of Simulations of Intrinsically Disordered Proteins
- PMID: 37463458
- PMCID: PMC11188088
- DOI: 10.1021/acs.jctc.3c00190
SOURSOP: A Python Package for the Analysis of Simulations of Intrinsically Disordered Proteins
Abstract
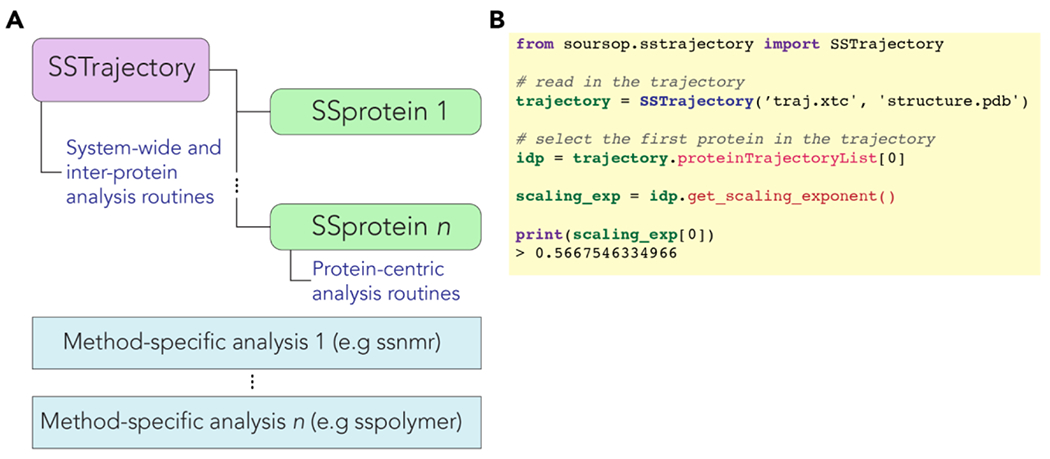
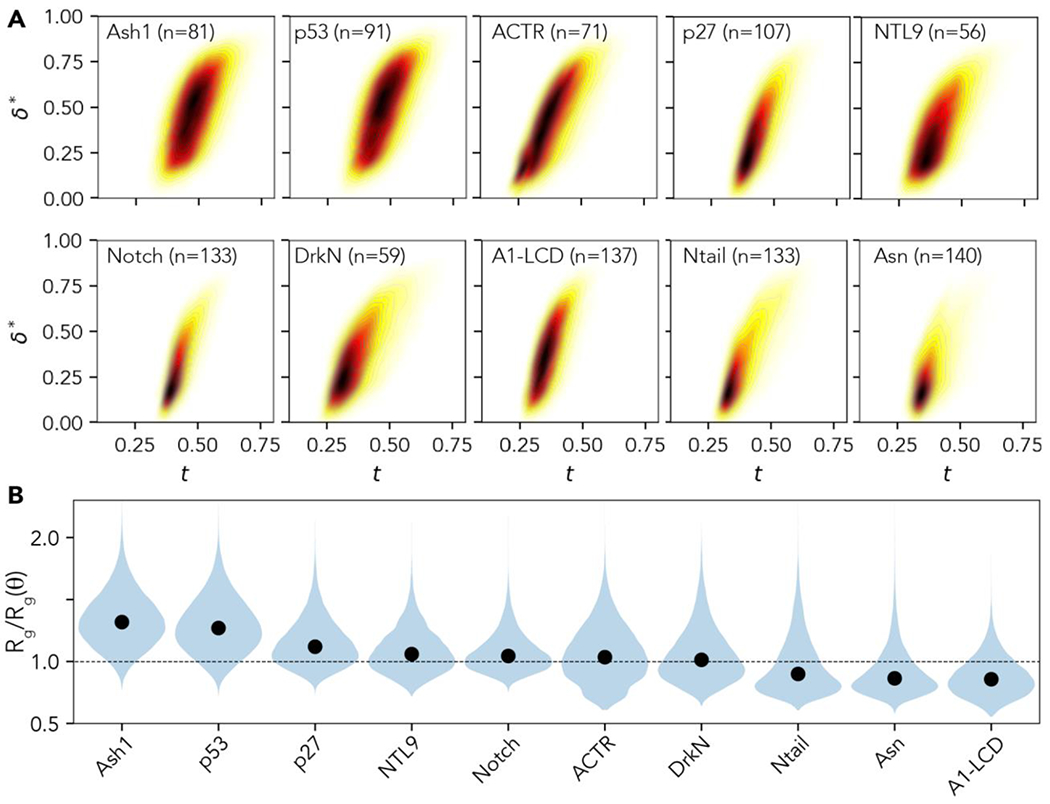
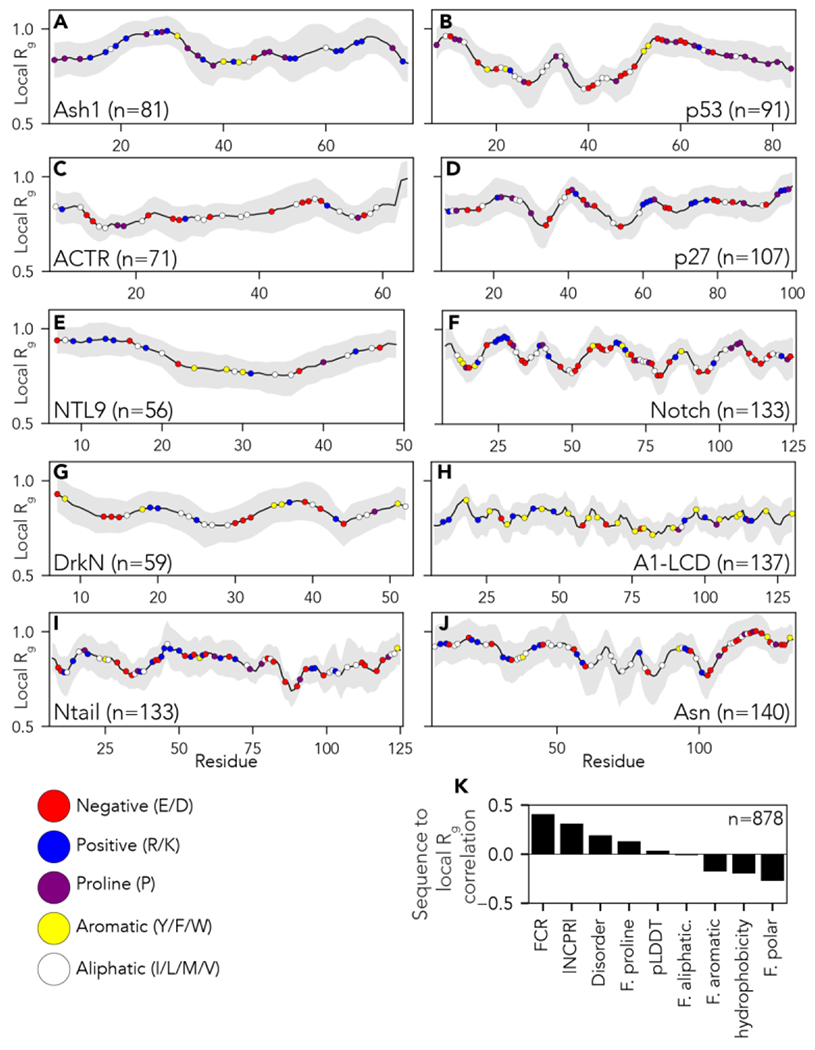
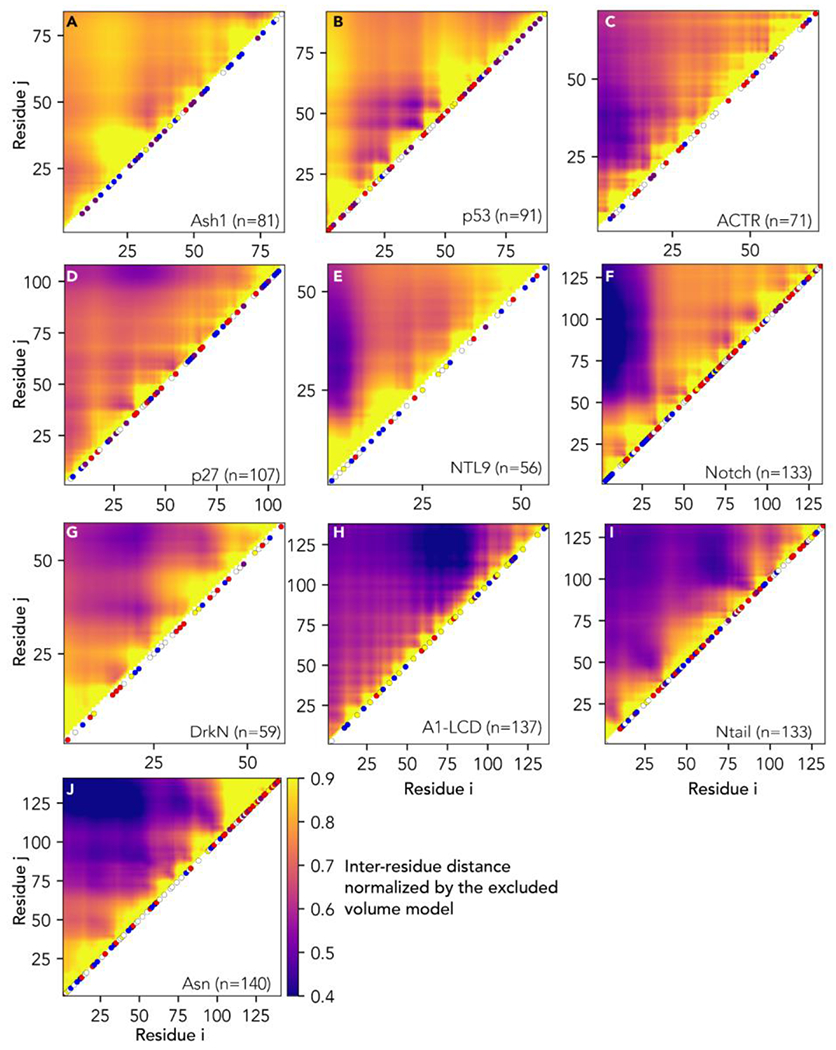
Conformational heterogeneity is a defining hallmark of intrinsically disordered proteins and protein regions (IDRs). The functions of IDRs and the emergent cellular phenotypes they control are associated with sequence-specific conformational ensembles. Simulations of conformational ensembles that are based on atomistic and coarse-grained models are routinely used to uncover the sequence-specific interactions that may contribute to IDR functions. These simulations are performed either independently or in conjunction with data from experiments. Functionally relevant features of IDRs can span a range of length scales. Extracting these features requires analysis routines that quantify a range of properties. Here, we describe a new analysis suite simulation analysis of unfolded regions of proteins (SOURSOP), an object-oriented and open-source toolkit designed for the analysis of simulated conformational ensembles of IDRs. SOURSOP implements several analysis routines motivated by principles in polymer physics, offering a unique collection of simple-to-use functions to characterize IDR ensembles. As an extendable framework, SOURSOP supports the development and implementation of new analysis routines that can be easily packaged and shared.
Figures
Update of
-
SOURSOP: A Python package for the analysis of simulations of intrinsically disordered proteins.bioRxiv [Preprint]. 2023 Feb 17:2023.02.16.528879. doi: 10.1101/2023.02.16.528879. bioRxiv. 2023. Update in: J Chem Theory Comput. 2023 Aug 22;19(16):5609-5620. doi: 10.1021/acs.jctc.3c00190 PMID: 36824878 Free PMC article. Updated. Preprint.
Similar articles
-
SOURSOP: A Python package for the analysis of simulations of intrinsically disordered proteins.bioRxiv [Preprint]. 2023 Feb 17:2023.02.16.528879. doi: 10.1101/2023.02.16.528879. bioRxiv. 2023. Update in: J Chem Theory Comput. 2023 Aug 22;19(16):5609-5620. doi: 10.1021/acs.jctc.3c00190 PMID: 36824878 Free PMC article. Updated. Preprint.
-
Expansion of Intrinsically Disordered Proteins Increases the Range of Stability of Liquid-Liquid Phase Separation.Molecules. 2020 Oct 15;25(20):4705. doi: 10.3390/molecules25204705. Molecules. 2020. PMID: 33076213 Free PMC article.
-
Sequence-to-Conformation Relationships of Disordered Regions Tethered to Folded Domains of Proteins.J Mol Biol. 2018 Aug 3;430(16):2403-2421. doi: 10.1016/j.jmb.2018.05.012. Epub 2018 May 12. J Mol Biol. 2018. PMID: 29763584
-
Predicting Conformational Ensembles of Intrinsically Disordered Proteins: From Molecular Dynamics to Machine Learning.J Phys Chem Lett. 2024 Aug 15;15(32):8177-8186. doi: 10.1021/acs.jpclett.4c01544. Epub 2024 Aug 2. J Phys Chem Lett. 2024. PMID: 39093570 Review.
-
Features of molecular recognition of intrinsically disordered proteins via coupled folding and binding.Protein Sci. 2019 Nov;28(11):1952-1965. doi: 10.1002/pro.3718. Epub 2019 Sep 4. Protein Sci. 2019. PMID: 31441158 Free PMC article. Review.
Cited by
-
Phosphorylation of disordered proteins tunes local and global intramolecular interactions.bioRxiv [Preprint]. 2024 Jun 12:2024.06.10.598315. doi: 10.1101/2024.06.10.598315. bioRxiv. 2024. Update in: Biophys J. 2024 Dec 3;123(23):4082-4096. doi: 10.1016/j.bpj.2024.10.021 PMID: 38915510 Free PMC article. Updated. Preprint.
-
Direct prediction of intrinsically disordered protein conformational properties from sequence.Nat Methods. 2024 Mar;21(3):465-476. doi: 10.1038/s41592-023-02159-5. Epub 2024 Jan 31. Nat Methods. 2024. PMID: 38297184 Free PMC article.
-
Aberrant phase separation is a common killing strategy of positively charged peptides in biology and human disease.bioRxiv [Preprint]. 2023 Mar 9:2023.03.09.531820. doi: 10.1101/2023.03.09.531820. bioRxiv. 2023. PMID: 36945394 Free PMC article. Preprint.
-
The Analytical Flory Random Coil Is a Simple-to-Use Reference Model for Unfolded and Disordered Proteins.J Phys Chem B. 2023 Jun 1;127(21):4746-4760. doi: 10.1021/acs.jpcb.3c01619. Epub 2023 May 18. J Phys Chem B. 2023. PMID: 37200094 Free PMC article.
-
Structural dynamics of the intrinsically disordered linker region of cardiac troponin T.bioRxiv [Preprint]. 2024 Oct 14:2024.05.30.596451. doi: 10.1101/2024.05.30.596451. bioRxiv. 2024. PMID: 38853835 Free PMC article. Preprint.
References
-
- van der Lee R; Buljan M; Lang B; Weatheritt RJ; Daughdrill GW; Dunker AK; Fuxreiter M; Gough J; Gsponer J; Jones DT; Kim PM; Kriwacki RW; Oldfield CJ; Pappu RV; Tompa P; Uversky VN; Wright PE; Babu MM Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev 2014, 114 (13), 6589–6631. - PMC - PubMed
-
- Sigler PB Acid Blobs & Negative Noodles. Nature 1988, 333, 210–212. - PubMed
-
- Ptitsyn OB; Uversky VN The Molten Globule Is a Third Thermodynamical State of Protein Molecules. FEBS Lett. 1994, 341 (1), 15–18. - PubMed
-
- Wright PE; Dyson HJ Intrinsically Unstructured Proteins: Re-Assessing the Protein Structure-Function Paradigm. J. Mol. Biol 1999, 293 (2), 321–331. - PubMed
-
- Babu MM; Kriwacki RW; Pappu RV Structural Biology. Versatility from Protein Disorder. Science 2012, 337 (6101), 1460–1461. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials