

Update on Autophagy Inhibitors in Cancer: Opening up to a Therapeutic Combination with Immune Checkpoint Inhibitors
- PMID: 37443736
- PMCID: PMC10341243
- DOI: 10.3390/cells12131702
Update on Autophagy Inhibitors in Cancer: Opening up to a Therapeutic Combination with Immune Checkpoint Inhibitors
Abstract
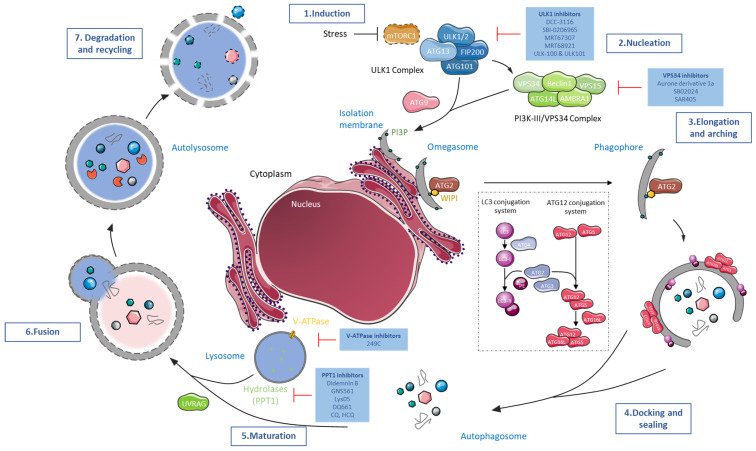
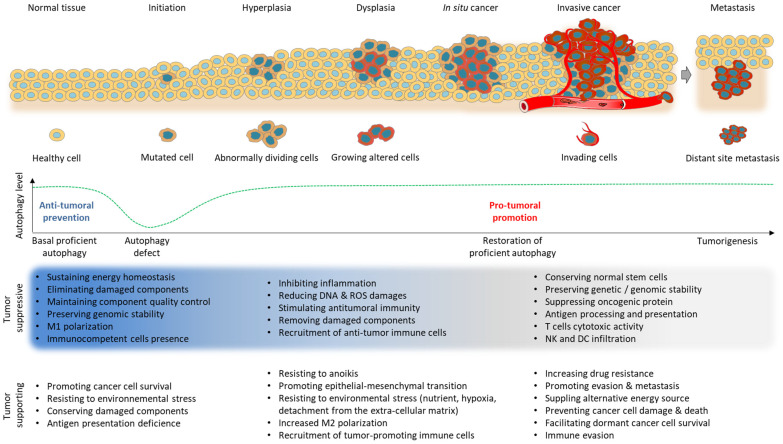
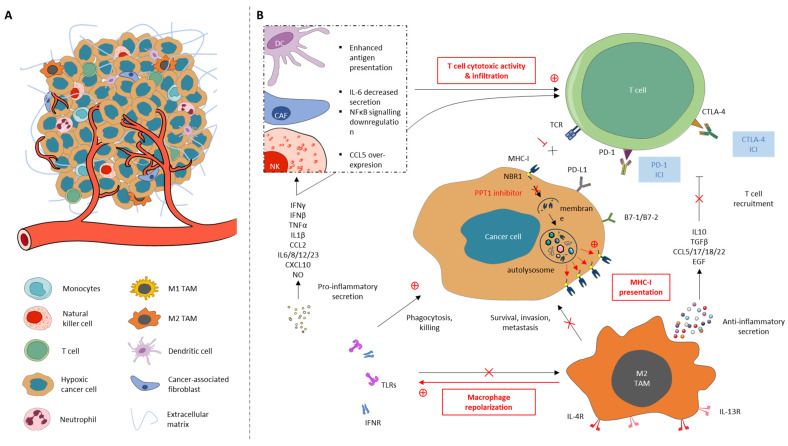
Autophagy is a highly conserved and natural degradation process that helps maintain cell homeostasis through the elimination of old, worn, and defective cellular components, ensuring proper cell energy intake. The degradative pathway constitutes a protective barrier against diverse human diseases including cancer. Autophagy basal level has been reported to be completely dysregulated during the entire oncogenic process. Autophagy influences not only cancer initiation, development, and maintenance but also regulates cancer response to therapy. Currently, autophagy inhibitor candidates mainly target the early autophagy process without any successful preclinical/clinical development. Lessons learned from autophagy pharmaceutical manipulation as a curative option progressively help to improve drug design and to encounter new targets of interest. Combinatorial strategies with autophagy modulators are supported by abundant evidence, especially dealing with immune checkpoint inhibitors, for which encouraging preclinical results have been recently published. GNS561, a PPT1 inhibitor, is a promising autophagy modulator as it has started a phase 2 clinical trial in liver cancer indication, combined with atezolizumab and bevacizumab, an assessment without precedent in the field. This approach paves a new road, leading to the resurgence of anticancer autophagy inhibitors as an attractive therapeutic target in cancer.
Keywords: PPT1; autophagy; cancer; clinical trial; combinational therapy; drug inhibitor.
Conflict of interest statement
The authors are employees of Genoscience Pharma.
Figures
Similar articles
-
GNS561, a clinical-stage PPT1 inhibitor, is efficient against hepatocellular carcinoma via modulation of lysosomal functions.Autophagy. 2022 Mar;18(3):678-694. doi: 10.1080/15548627.2021.1988357. Epub 2021 Nov 5. Autophagy. 2022. PMID: 34740311 Free PMC article.
-
Autophagy modulation: a target for cancer treatment development.Cancer Chemother Pharmacol. 2015 Mar;75(3):439-47. doi: 10.1007/s00280-014-2637-z. Epub 2014 Nov 25. Cancer Chemother Pharmacol. 2015. PMID: 25422156 Review.
-
Cancer Immunotherapy - Immune Checkpoint Inhibitors in Hepatocellular Carcinoma.Recent Pat Anticancer Drug Discov. 2021;16(2):239-248. doi: 10.2174/1574892816666210212145107. Recent Pat Anticancer Drug Discov. 2021. PMID: 33583384 Review.
-
Systemic treatments for metastatic cutaneous melanoma.Cochrane Database Syst Rev. 2018 Feb 6;2(2):CD011123. doi: 10.1002/14651858.CD011123.pub2. Cochrane Database Syst Rev. 2018. PMID: 29405038 Free PMC article. Review.
-
Pharmacological inhibitors of autophagy as novel cancer therapeutic agents.Pharmacol Res. 2016 Mar;105:164-75. doi: 10.1016/j.phrs.2016.01.028. Epub 2016 Jan 28. Pharmacol Res. 2016. PMID: 26826398 Review.
Cited by
-
A review of chemotherapeutic drugs-induced arrhythmia and potential intervention with traditional Chinese medicines.Front Pharmacol. 2024 Mar 20;15:1340855. doi: 10.3389/fphar.2024.1340855. eCollection 2024. Front Pharmacol. 2024. PMID: 38572424 Free PMC article. Review.
-
Advances of Protein Palmitoylation in Tumor Cell Deaths.Cancers (Basel). 2023 Nov 21;15(23):5503. doi: 10.3390/cancers15235503. Cancers (Basel). 2023. PMID: 38067206 Free PMC article. Review.
-
Targeting autophagy can synergize the efficacy of immune checkpoint inhibitors against therapeutic resistance: New promising strategy to reinvigorate cancer therapy.Heliyon. 2024 Sep 3;10(18):e37376. doi: 10.1016/j.heliyon.2024.e37376. eCollection 2024 Sep 30. Heliyon. 2024. PMID: 39309904 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
