

Stochastic motion and transcriptional dynamics of pairs of distal DNA loci on a compacted chromosome
- PMID: 37384691
- PMCID: PMC10439308
- DOI: 10.1126/science.adf5568
Stochastic motion and transcriptional dynamics of pairs of distal DNA loci on a compacted chromosome
Abstract
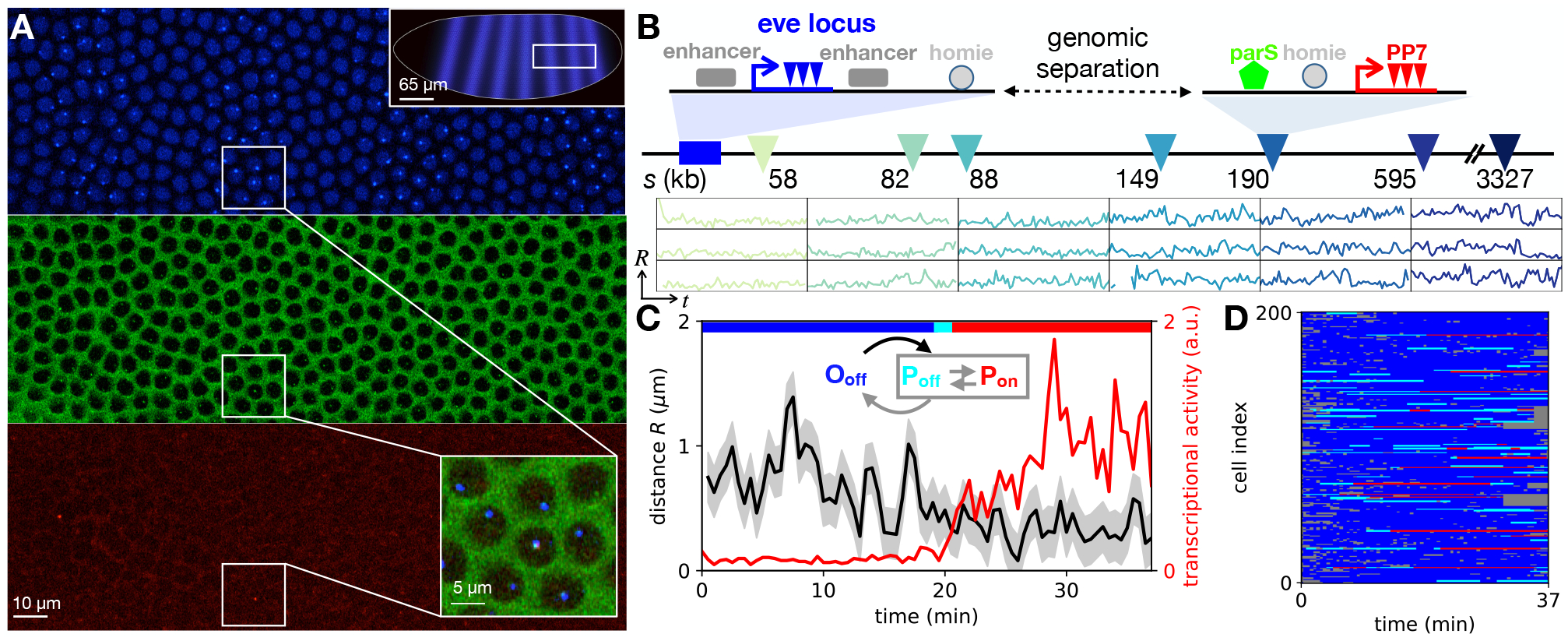
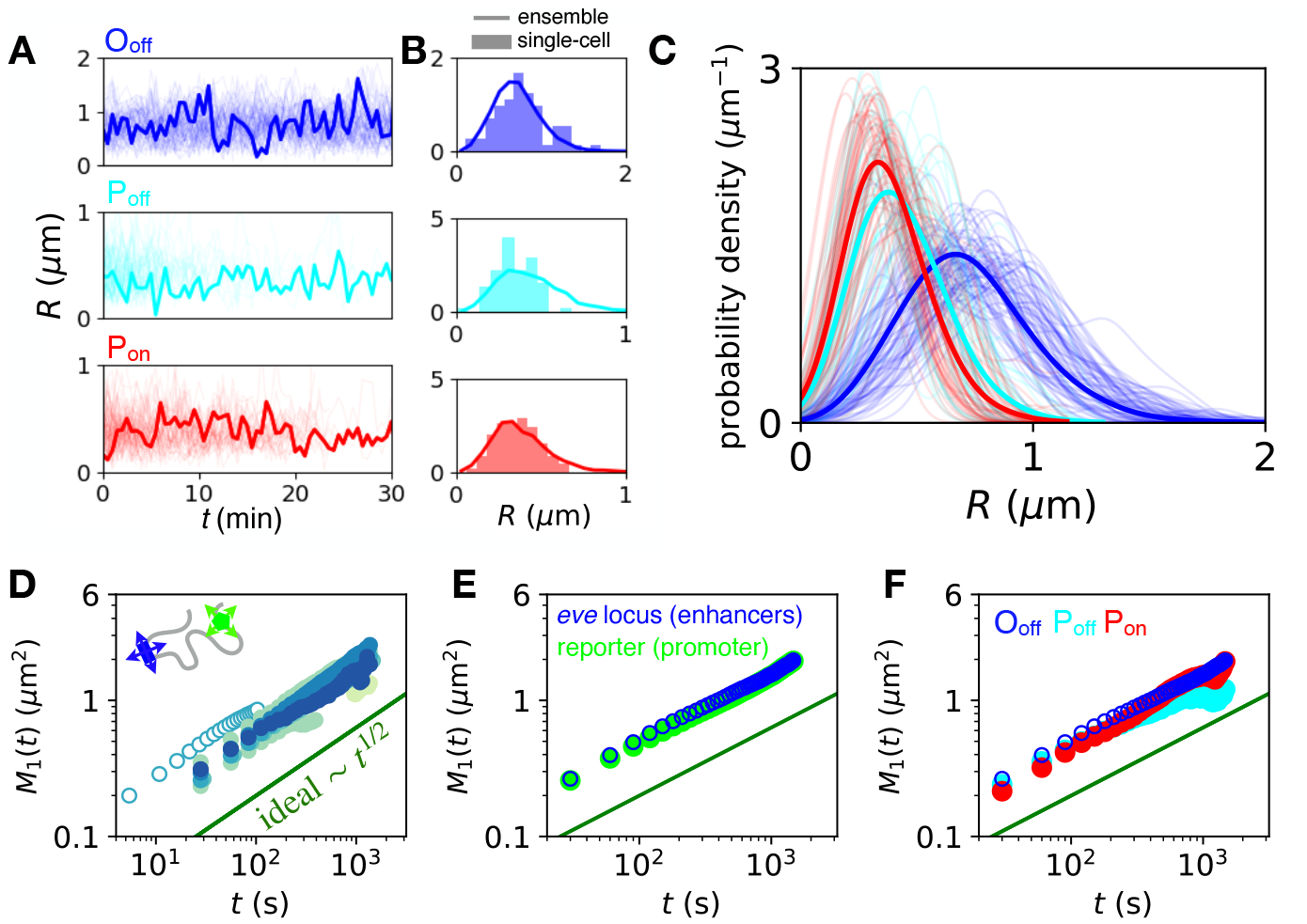
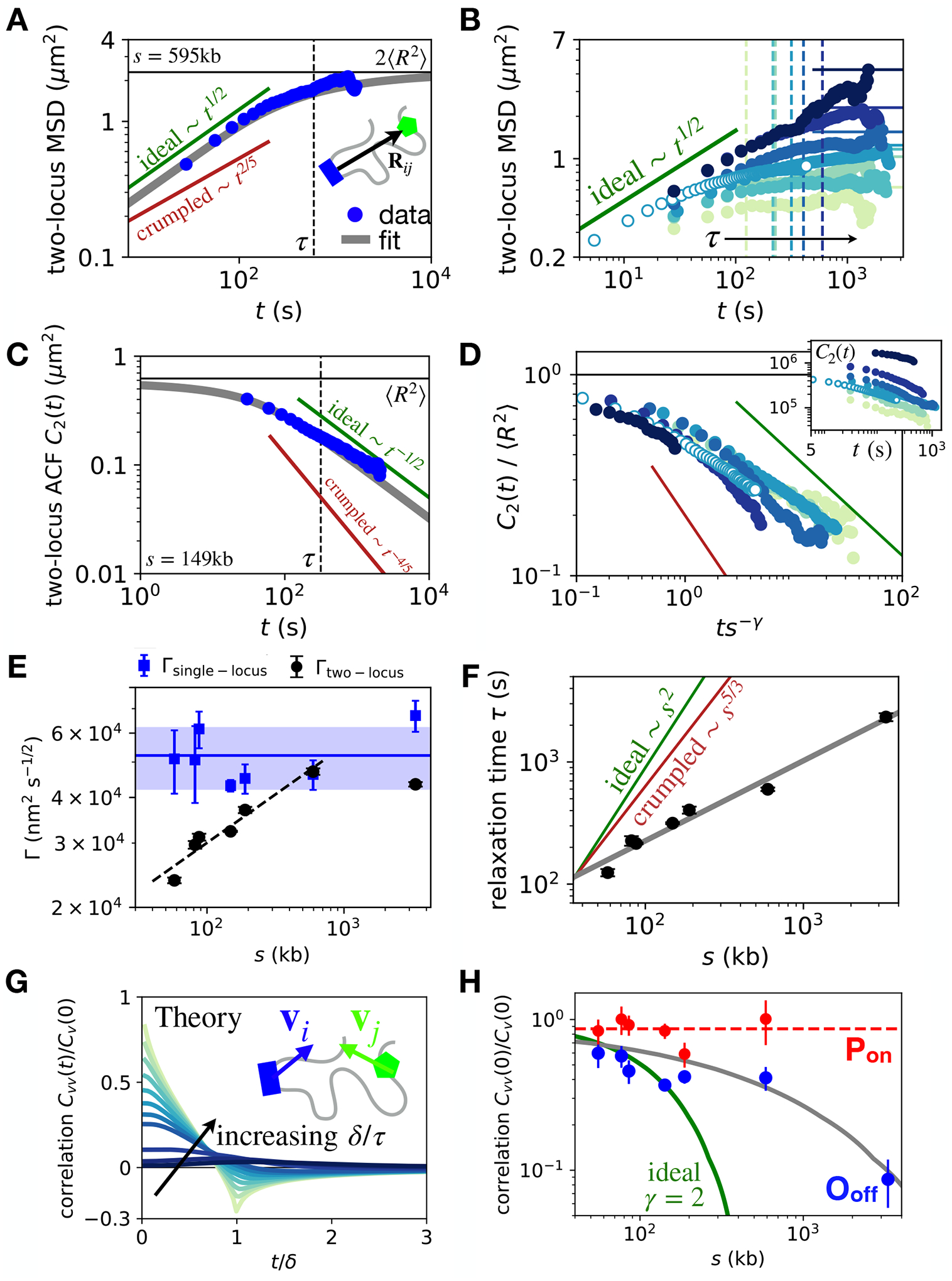
Chromosomes in the eukaryotic nucleus are highly compacted. However, for many functional processes, including transcription initiation, the pairwise motion of distal chromosomal elements such as enhancers and promoters is essential and necessitates dynamic fluidity. Here, we used a live-imaging assay to simultaneously measure the positions of pairs of enhancers and promoters and their transcriptional output while systematically varying the genomic separation between these two DNA loci. Our analysis reveals the coexistence of a compact globular organization and fast subdiffusive dynamics. These combined features cause an anomalous scaling of polymer relaxation times with genomic separation leading to long-ranged correlations. Thus, encounter times of DNA loci are much less dependent on genomic distance than predicted by existing polymer models, with potential consequences for eukaryotic gene expression.
Conflict of interest statement
Figures

Update of
-
Stochastic motion and transcriptional dynamics of pairs of distal DNA loci on a compacted chromosome.bioRxiv [Preprint]. 2023 Feb 13:2023.01.18.524527. doi: 10.1101/2023.01.18.524527. bioRxiv. 2023. Update in: Science. 2023 Jun 30;380(6652):1357-1362. doi: 10.1126/science.adf5568. PMID: 36711618 Free PMC article. Updated. Preprint.
Similar articles
-
Stochastic motion and transcriptional dynamics of pairs of distal DNA loci on a compacted chromosome.bioRxiv [Preprint]. 2023 Feb 13:2023.01.18.524527. doi: 10.1101/2023.01.18.524527. bioRxiv. 2023. Update in: Science. 2023 Jun 30;380(6652):1357-1362. doi: 10.1126/science.adf5568. PMID: 36711618 Free PMC article. Updated. Preprint.
-
Diffusing polymers in confined microdomains and estimation of chromosomal territory sizes from chromosome capture data.Phys Rev Lett. 2013 Jun 14;110(24):248105. doi: 10.1103/PhysRevLett.110.248105. Epub 2013 Jun 14. Phys Rev Lett. 2013. PMID: 25165966
-
Visualizing the Role of Boundary Elements in Enhancer-Promoter Communication.Mol Cell. 2020 Apr 16;78(2):224-235.e5. doi: 10.1016/j.molcel.2020.02.007. Epub 2020 Feb 27. Mol Cell. 2020. PMID: 32109364
-
Dynamics of transcriptional enhancers and chromosome topology in gene regulation.Dev Growth Differ. 2019 Jun;61(5):343-352. doi: 10.1111/dgd.12597. Epub 2019 Feb 19. Dev Growth Differ. 2019. PMID: 30780195 Free PMC article. Review.
-
Are TADs supercoiled?Nucleic Acids Res. 2019 Jan 25;47(2):521-532. doi: 10.1093/nar/gky1091. Nucleic Acids Res. 2019. PMID: 30395328 Free PMC article. Review.
Cited by
-
An extrinsic motor directs chromatin loop formation by cohesin.EMBO J. 2024 Oct;43(19):4173-4196. doi: 10.1038/s44318-024-00202-5. Epub 2024 Aug 19. EMBO J. 2024. PMID: 39160275 Free PMC article.
-
Node features of chromosome structure networks and their connections to genome annotation.Comput Struct Biotechnol J. 2024 May 17;23:2240-2250. doi: 10.1016/j.csbj.2024.05.026. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 38827231 Free PMC article.
-
Strong interactions between highly dynamic lamina-associated domains and the nuclear envelope stabilize the 3D architecture of Drosophila interphase chromatin.Epigenetics Chromatin. 2023 May 30;16(1):21. doi: 10.1186/s13072-023-00492-9. Epigenetics Chromatin. 2023. PMID: 37254161 Free PMC article.
-
Spatial and temporal organization of the genome: Current state and future aims of the 4D nucleome project.Mol Cell. 2023 Aug 3;83(15):2624-2640. doi: 10.1016/j.molcel.2023.06.018. Epub 2023 Jul 6. Mol Cell. 2023. PMID: 37419111 Free PMC article. Review.
-
Activity-driven chromatin organization during interphase: Compaction, segregation, and entanglement suppression.Proc Natl Acad Sci U S A. 2024 May 21;121(21):e2401494121. doi: 10.1073/pnas.2401494121. Epub 2024 May 16. Proc Natl Acad Sci U S A. 2024. PMID: 38753513 Free PMC article.
References
-
- Walther. Flemming. Zellsubstanz, Kern Und Zelltheilung. Vogel, Leipzig, 1882.
-
- Milo R and Phillips R. Cell Biology by the Numbers. CRC Press, 2015.
-
- Stadhouders Ralph, Filion Guillaume J, and Graf Thomas. Transcription factors and 3D genome conformation in cell-fate decisions. Nature, 569(7756):345–354, 2019. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
