

Non-Vesicular Release of Alarmin Prothymosin α Complex Associated with Annexin-2 Flop-Out
- PMID: 37371039
- PMCID: PMC10296757
- DOI: 10.3390/cells12121569
Non-Vesicular Release of Alarmin Prothymosin α Complex Associated with Annexin-2 Flop-Out
Abstract
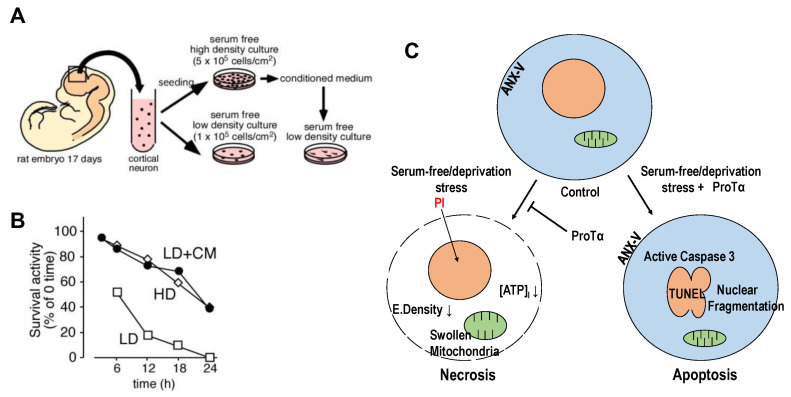
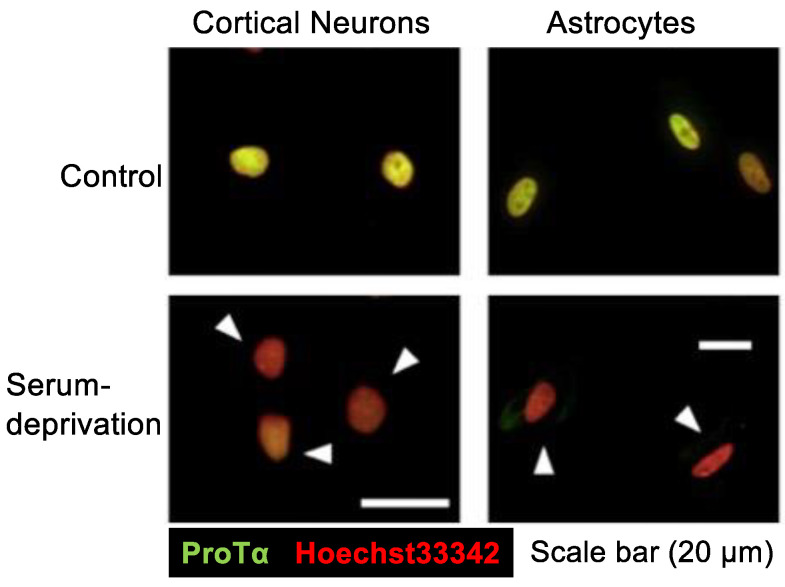
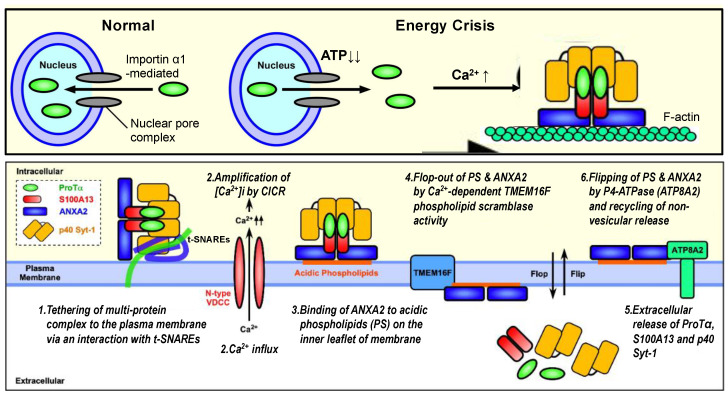
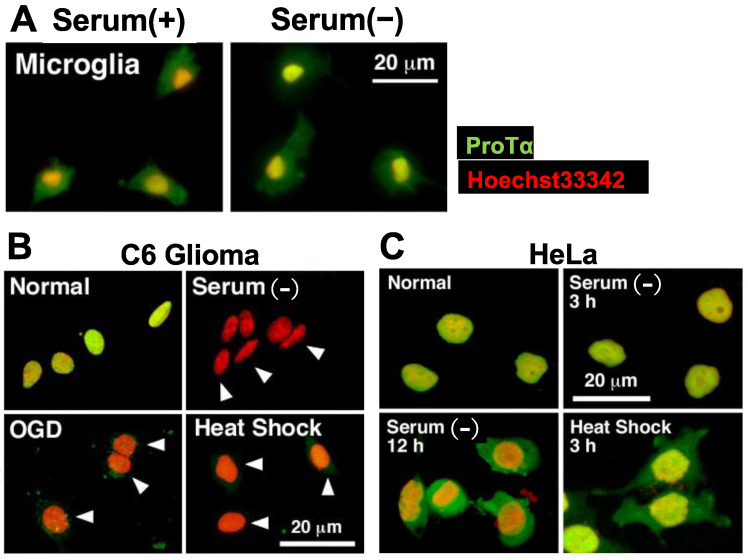
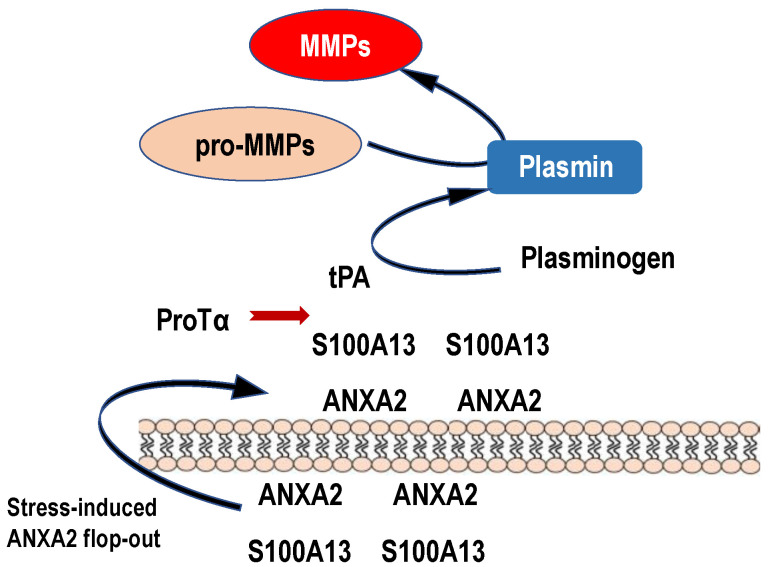
Nuclear protein prothymosin α (ProTα) is a unique member of damage-associated molecular patterns (DAMPs)/alarmins. ProTα prevents neuronal necrosis by causing a cell death mode switch in serum-starving or ischemic/reperfusion models in vitro and in vivo. Underlying receptor mechanisms include Toll-like receptor 4 (TLR4) and Gi-coupled receptor. Recent studies have revealed that the mode of the fatal stress-induced extracellular release of nuclear ProTα from cortical neurons in primary cultures, astrocytes and C6 glioma cells has two steps: ATP loss-induced nuclear release and the Ca2+-mediated formation of a multiple protein complex and its extracellular release. Under the serum-starving condition, ProTα is diffused from the nucleus throughout the cell due to the ATP loss-induced impairment of importin α-mediated nuclear transport. Subsequent mechanisms are all Ca2+-dependent. They include the formation of a protein complex with ProTα, S100A13, p40 Syt-1 and Annexin A2 (ANXA2); the fusion of the protein complex to the plasma membrane via p40 Syt-1-Stx-1 interaction; and TMEM16F scramblase-mediated ANXA2 flop-out. Subsequently, the protein complex is extracellularly released, leaving ANXA2 on the outer cell surface. The ANXA2 is then flipped in by a force of ATP8A2 activity, and the non-vesicular release of protein complex is repeated. Thus, the ANXA2 flop-out could play key roles in a new type of non-vesicular and non-classical release for DAMPs/alarmins, which is distinct from the modes conducted via gasdermin D or mixed-lineage kinase domain-like pseudokinase pores.
Keywords: DAMPs; GSDMD; MLKL; S100A13; SNARE complex; alarmins; exosomes; flippase; scramblase.
Conflict of interest statement
The author Hiroshi Ueda declares that he has no conflict of interest.
Figures
Similar articles
-
Annexin A2 Flop-Out Mediates the Non-Vesicular Release of DAMPs/Alarmins from C6 Glioma Cells Induced by Serum-Free Conditions.Cells. 2021 Mar 5;10(3):567. doi: 10.3390/cells10030567. Cells. 2021. PMID: 33807671 Free PMC article.
-
Involvement of SNARE Protein Interaction for Non-classical Release of DAMPs/Alarmins Proteins, Prothymosin Alpha and S100A13.Cell Mol Neurobiol. 2021 Nov;41(8):1817-1828. doi: 10.1007/s10571-020-00950-y. Epub 2020 Aug 27. Cell Mol Neurobiol. 2021. PMID: 32856232
-
Stress-induced non-vesicular release of prothymosin-α initiated by an interaction with S100A13, and its blockade by caspase-3 cleavage.Cell Death Differ. 2010 Nov;17(11):1760-72. doi: 10.1038/cdd.2010.52. Epub 2010 May 14. Cell Death Differ. 2010. PMID: 20467443
-
Prothymosin Alpha: An Alarmin and More..Curr Med Chem. 2017;24(17):1747-1760. doi: 10.2174/0929867324666170518110033. Curr Med Chem. 2017. PMID: 28521686 Review.
-
Prothymosin α Plays Role as a Brain Guardian through Ecto-F1 ATPase-P2Y12 Complex and TLR4/MD2.Cells. 2023 Feb 2;12(3):496. doi: 10.3390/cells12030496. Cells. 2023. PMID: 36766838 Free PMC article. Review.
Cited by
-
Effect of prothymosin α on neuroplasticity following cerebral ischemia‑reperfusion injury.Mol Med Rep. 2024 Apr;29(4):59. doi: 10.3892/mmr.2024.13183. Epub 2024 Feb 23. Mol Med Rep. 2024. PMID: 38391118 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
