

Molecular mechanisms of tumor resistance to radiotherapy
- PMID: 37322433
- PMCID: PMC10268375
- DOI: 10.1186/s12943-023-01801-2
Molecular mechanisms of tumor resistance to radiotherapy
Abstract
Background: Cancer is the most prevalent cause of death globally, and radiotherapy is considered the standard of care for most solid tumors, including lung, breast, esophageal, and colorectal cancers and glioblastoma. Resistance to radiation can lead to local treatment failure and even cancer recurrence.
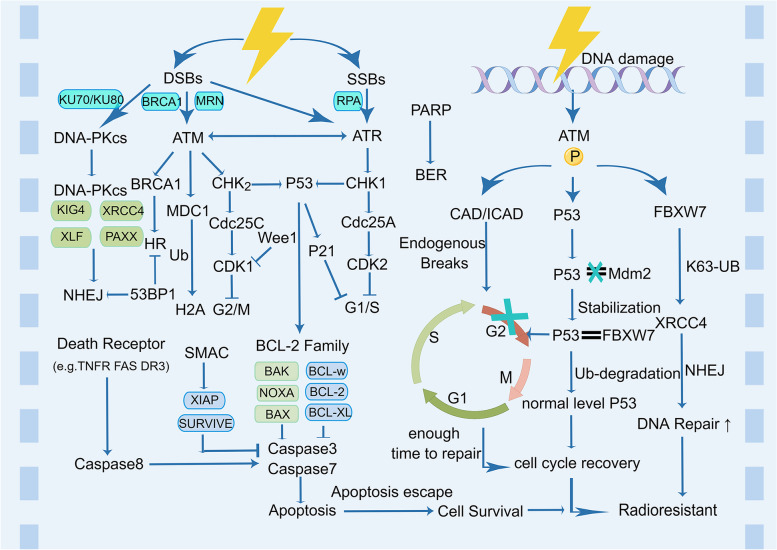
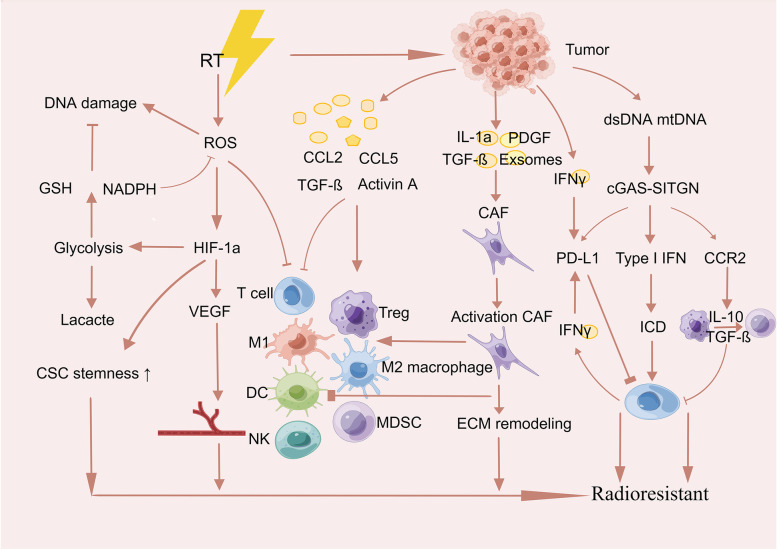
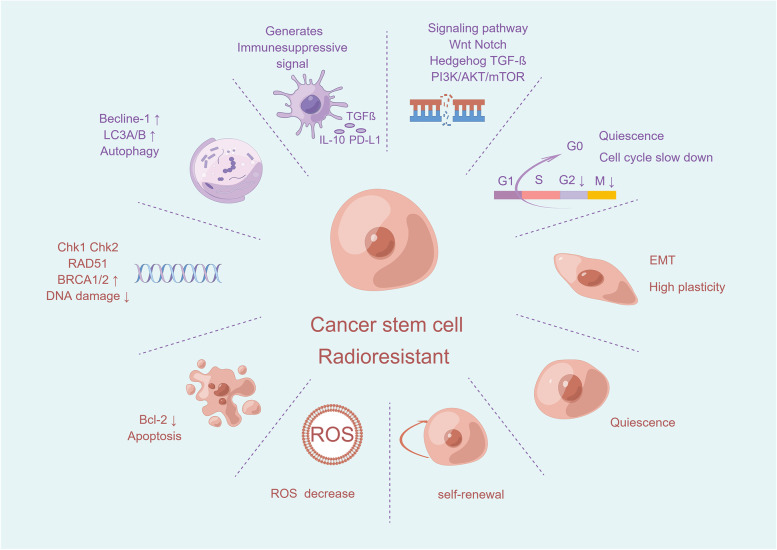
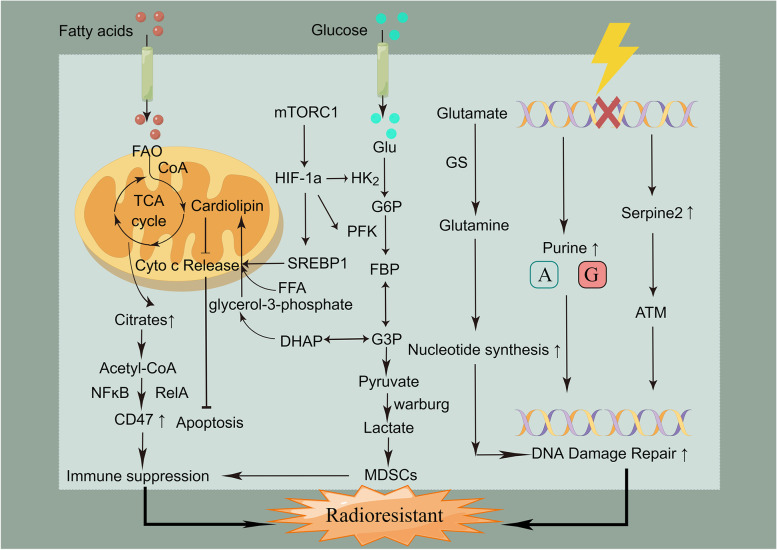
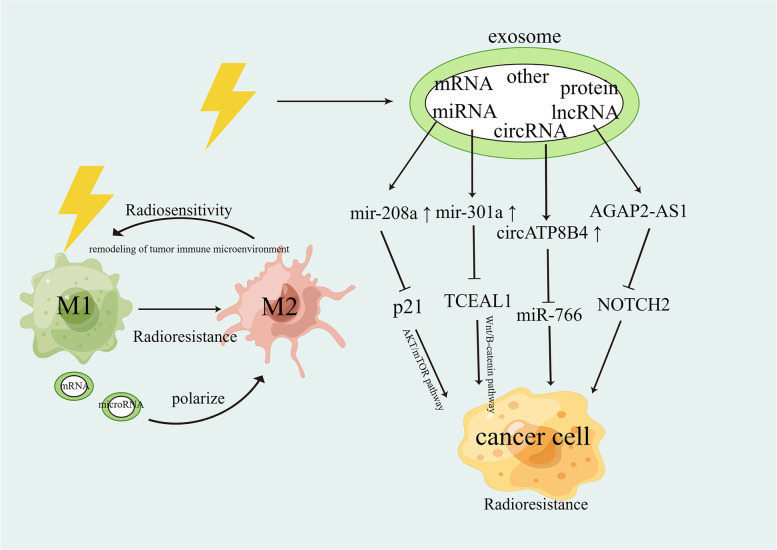
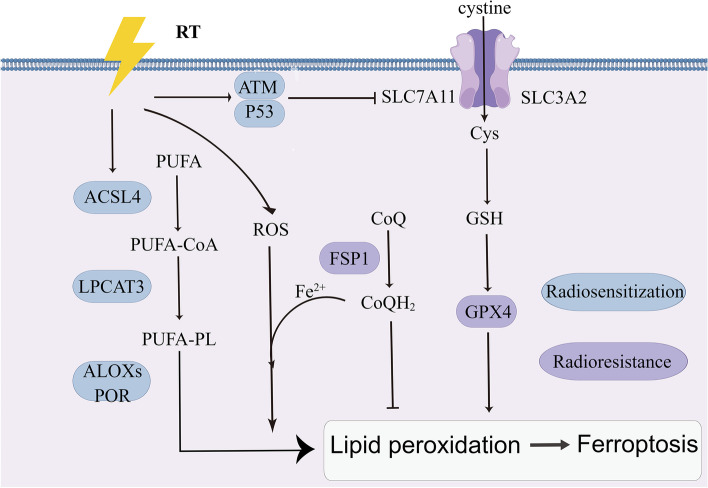
Main body: In this review, we have extensively discussed several crucial aspects that cause resistance of cancer to radiation therapy, including radiation-induced DNA damage repair, cell cycle arrest, apoptosis escape, abundance of cancer stem cells, modification of cancer cells and their microenvironment, presence of exosomal and non-coding RNA, metabolic reprogramming, and ferroptosis. We aim to focus on the molecular mechanisms of cancer radiotherapy resistance in relation to these aspects and to discuss possible targets to improve treatment outcomes.
Conclusions: Studying the molecular mechanisms responsible for radiotherapy resistance and its interactions with the tumor environment will help improve cancer responses to radiotherapy. Our review provides a foundation to identify and overcome the obstacles to effective radiotherapy.
Keywords: Cancer; Radiotherapy; Tumor resistance.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Radiotherapy resistance: identifying universal biomarkers for various human cancers.J Cancer Res Clin Oncol. 2022 May;148(5):1015-1031. doi: 10.1007/s00432-022-03923-4. Epub 2022 Feb 3. J Cancer Res Clin Oncol. 2022. PMID: 35113235 Review.
-
Coordination-driven FBXW7 DNAzyme-Fe nanoassembly enables a binary switch of breast cancer cell cycle checkpoint responses for enhanced ferroptosis-radiotherapy.Acta Biomater. 2023 Oct 1;169:434-450. doi: 10.1016/j.actbio.2023.07.042. Epub 2023 Jul 27. Acta Biomater. 2023. PMID: 37516418
-
Value of long non-coding RNA Rpph1 in esophageal cancer and its effect on cancer cell sensitivity to radiotherapy.World J Gastroenterol. 2020 Apr 21;26(15):1775-1791. doi: 10.3748/wjg.v26.i15.1775. World J Gastroenterol. 2020. PMID: 32351293 Free PMC article.
-
Review of possible mechanisms of radiotherapy resistance in cervical cancer.Front Oncol. 2023 Aug 22;13:1164985. doi: 10.3389/fonc.2023.1164985. eCollection 2023. Front Oncol. 2023. PMID: 37692844 Free PMC article. Review.
-
A DNA Repair and Cell-Cycle Gene Expression Signature in Primary and Recurrent Glioblastoma: Prognostic Value and Clinical Implications.Cancer Res. 2019 Mar 15;79(6):1226-1238. doi: 10.1158/0008-5472.CAN-18-2076. Epub 2019 Jan 23. Cancer Res. 2019. PMID: 30674534
Cited by
-
Radio-miRs: a comprehensive view of radioresistance-related microRNAs.Genetics. 2024 Aug 7;227(4):iyae097. doi: 10.1093/genetics/iyae097. Genetics. 2024. PMID: 38963803 Free PMC article. Review.
-
The role of RASA2 in predicting radioresistance in lung cancer through regulation of p53.Transl Lung Cancer Res. 2024 Mar 29;13(3):587-602. doi: 10.21037/tlcr-24-160. Epub 2024 Mar 27. Transl Lung Cancer Res. 2024. PMID: 38601440 Free PMC article.
-
A molecular classification system for estimating radiotherapy response and anticancer immunity for individual breast cancer patients.Front Oncol. 2023 Oct 20;13:1288698. doi: 10.3389/fonc.2023.1288698. eCollection 2023. Front Oncol. 2023. PMID: 37927478 Free PMC article.
-
New strategies for lung cancer diagnosis and treatment: applications and advances in nanotechnology.Biomark Res. 2024 Nov 13;12(1):136. doi: 10.1186/s40364-024-00686-7. Biomark Res. 2024. PMID: 39533445 Free PMC article. Review.
-
DNA methylation-regulated HK1 overexpression contributes to irradiation-resistance by promoting glycolysis in non-small cell lung cancer.Am J Cancer Res. 2024 Sep 15;14(9):4306-4319. doi: 10.62347/QMGJ2157. eCollection 2024. Am J Cancer Res. 2024. PMID: 39417179 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
