

A T Cell-Engaging Tumor Organoid Platform for Pancreatic Cancer Immunotherapy
- PMID: 37271874
- PMCID: PMC10427404
- DOI: 10.1002/advs.202300548
A T Cell-Engaging Tumor Organoid Platform for Pancreatic Cancer Immunotherapy
Abstract
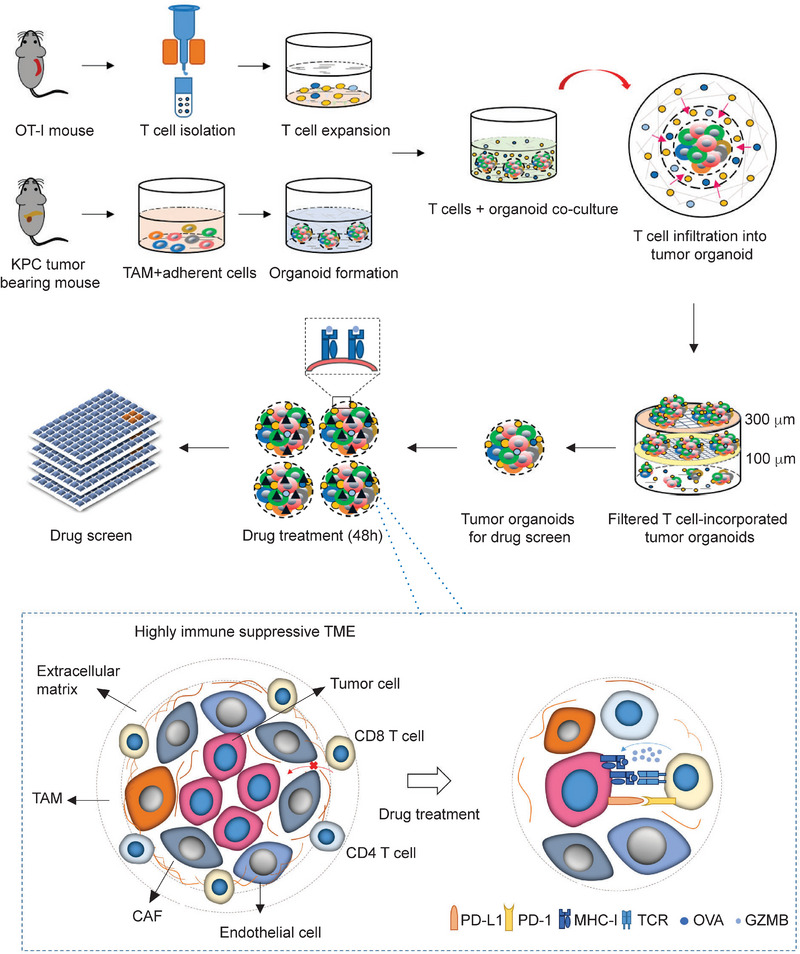
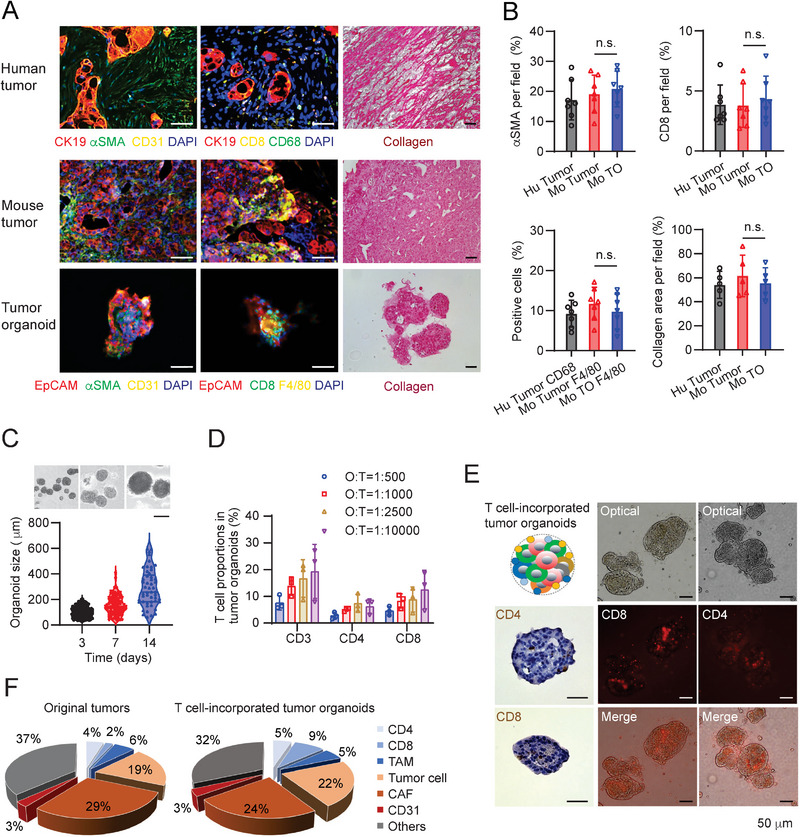
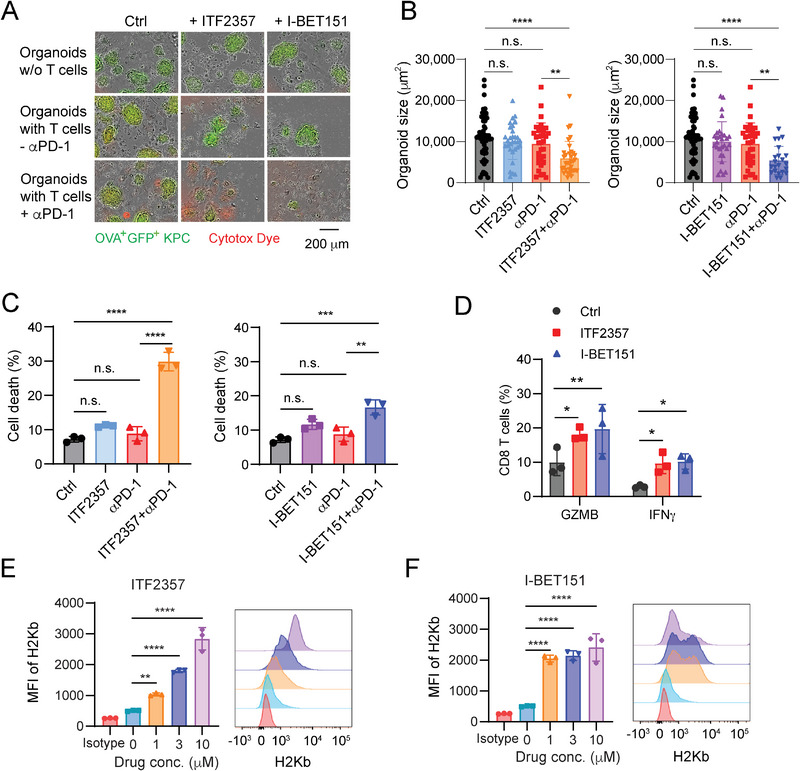
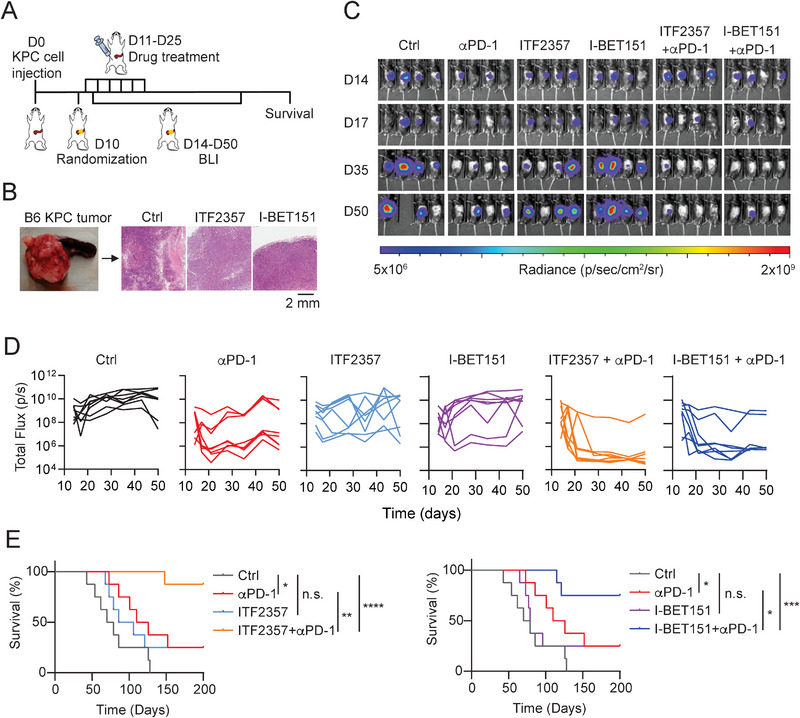
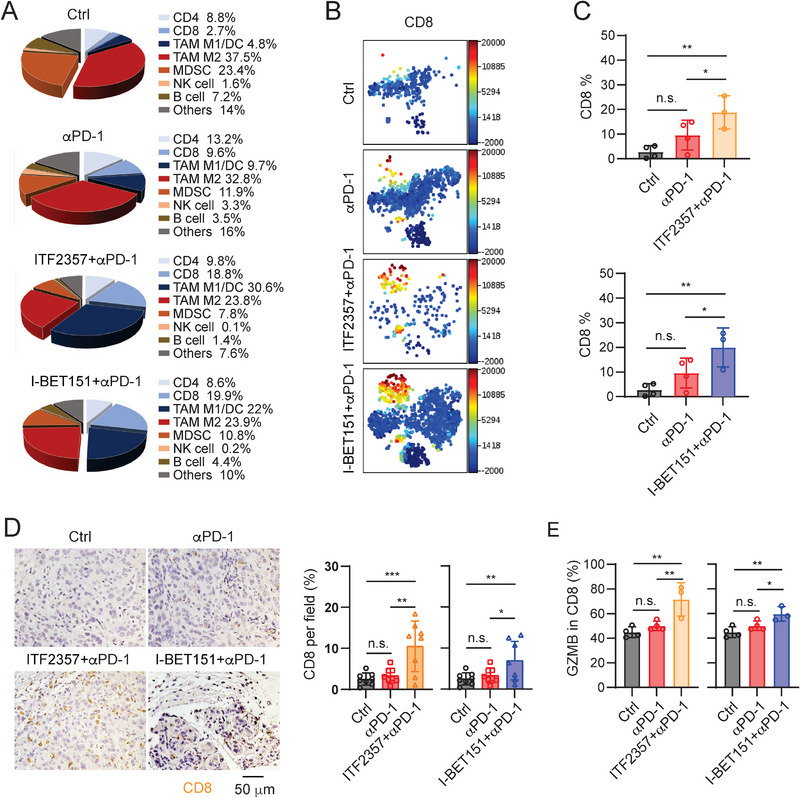
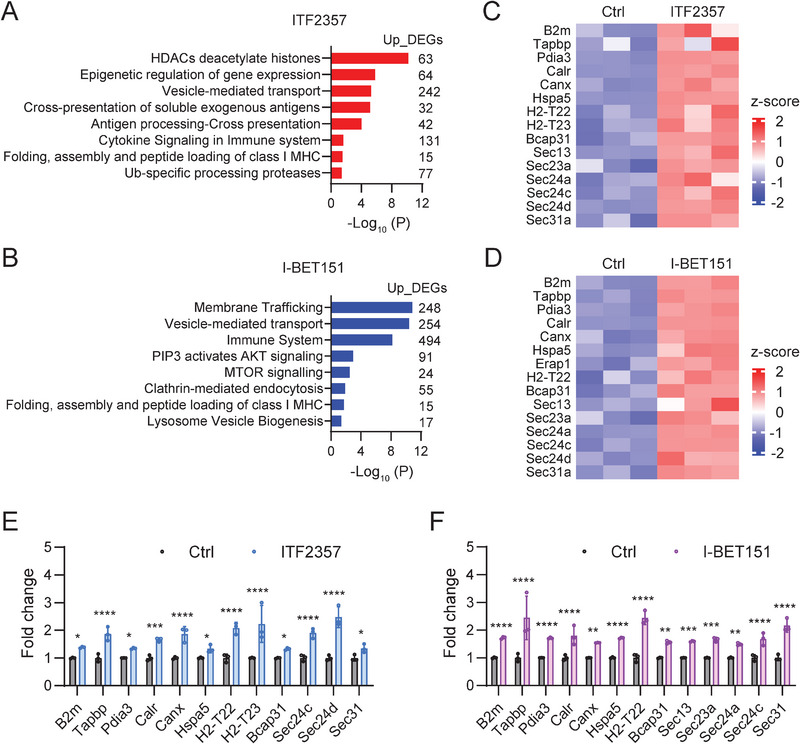
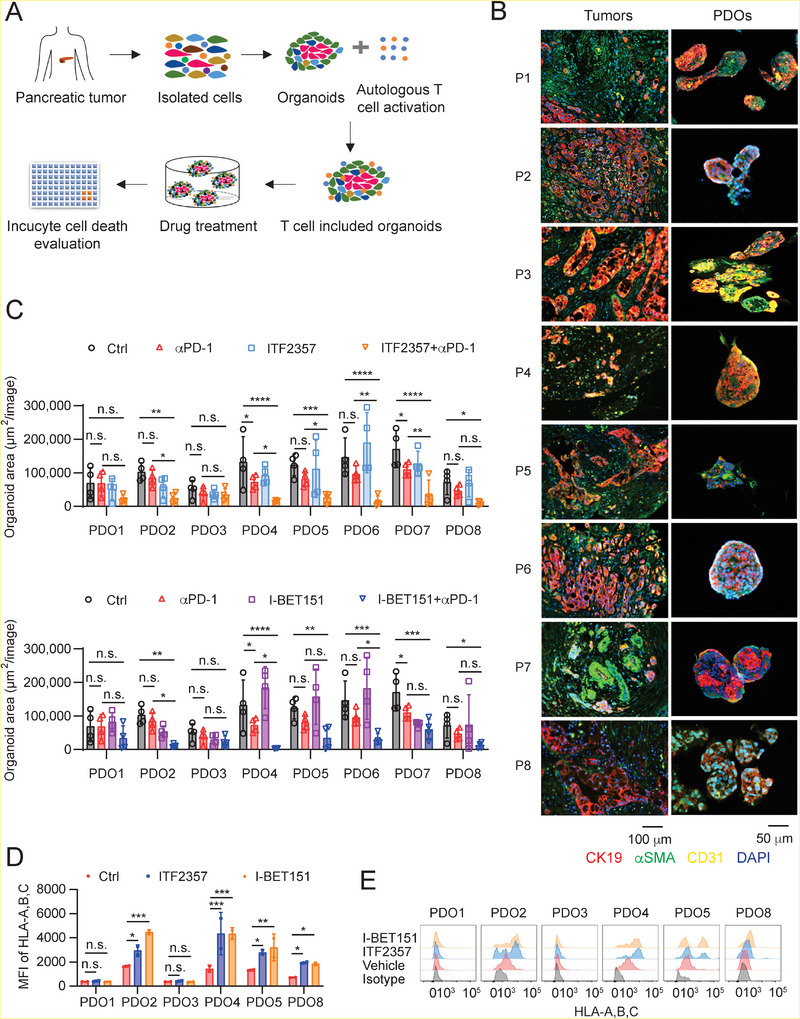
Pancreatic ductal adenocarcinoma (PDA) is a clinically challenging disease with limited treatment options. Despite a small percentage of cases with defective mismatch DNA repair (dMMR), PDA is included in the most immune-resistant cancer types that are poorly responsive to immune checkpoint blockade (ICB) therapy. To facilitate drug discovery combating this immunosuppressive tumor type, a high-throughput drug screen platform is established with the newly developed T cell-incorporated pancreatic tumor organoid model. Tumor-specific T cells are included in the pancreatic tumor organoids by two-step cell packaging, fully recapitulating immune infiltration in the immunosuppressive tumor microenvironment (TME). The organoids are generated with key components in the original tumor, including epithelial, vascular endothelial, fibroblast and macrophage cells, and then packaged with T cells into their outside layer mimicking a physical barrier and enabling T cell infiltration and cytotoxicity studies. In the PDA organoid-based screen, epigenetic inhibitors ITF2357 and I-BET151 are identified, which in combination with anti-PD-1 based therapy show considerably greater anti-tumor effect. The combinatorial treatment turns the TME from immunosuppressive to immunoactive, up-regulates the MHC-I antigen processing and presentation, and enhances the effector T cell activity. The standardized PDA organoid model has shown great promise to accelerate drug discovery for the immunosuppressive cancer.
Keywords: epigenetic inhibitors; high-throughput drug screen; immunotherapy; pancreatic ductal adenocarcinoma; patient-derived organoid; tumor antigen presentation; tumor organoid.
© 2023 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
M.O. received research funds from Eli Lilly and Bayer and has consulting roles with Pfizer, AstraZeneca, and Novartis. X.L. is a cofounder of Reactimm Therapeutics LLC. X.L. is an inventor on the pending patent PCT/US2021/062842, “Methods to sensitize cancer cells and standardized assay for immunotherapy”.
Figures

Similar articles
-
A nanodrug simultaneously inhibits pancreatic stellate cell activation and regulatory T cell infiltration to promote the immunotherapy of pancreatic cancer.Acta Biomater. 2023 Oct 1;169:451-463. doi: 10.1016/j.actbio.2023.08.007. Epub 2023 Aug 10. Acta Biomater. 2023. PMID: 37572982
-
T-cell programming in pancreatic adenocarcinoma: a review.Cancer Gene Ther. 2017 Mar;24(3):106-113. doi: 10.1038/cgt.2016.66. Epub 2016 Dec 2. Cancer Gene Ther. 2017. PMID: 27910859 Review.
-
Combination immunotherapy for pancreatic cancer: challenges and future considerations.Expert Rev Clin Immunol. 2022 Nov;18(11):1173-1186. doi: 10.1080/1744666X.2022.2120471. Epub 2022 Sep 11. Expert Rev Clin Immunol. 2022. PMID: 36045547 Review.
-
Hollow Cu2MoS4 nanoparticles loaded with immune checkpoint inhibitors reshape the tumor microenvironment to enhance immunotherapy for pancreatic cancer.Acta Biomater. 2024 Jan 1;173:365-377. doi: 10.1016/j.actbio.2023.10.024. Epub 2023 Oct 26. Acta Biomater. 2024. PMID: 37890815
-
Cancer-initiating cells in human pancreatic cancer organoids are maintained by interactions with endothelial cells.Cancer Lett. 2021 Feb 1;498:42-53. doi: 10.1016/j.canlet.2020.10.012. Epub 2020 Nov 12. Cancer Lett. 2021. PMID: 33188841
Cited by
-
Organoids: new frontiers in tumor immune microenvironment research.Front Immunol. 2024 Jul 29;15:1422031. doi: 10.3389/fimmu.2024.1422031. eCollection 2024. Front Immunol. 2024. PMID: 39136020 Free PMC article. Review.
-
Advancing cancer research through organoid technology.J Transl Med. 2024 Nov 8;22(1):1007. doi: 10.1186/s12967-024-05824-1. J Transl Med. 2024. PMID: 39516934 Free PMC article. Review.
-
A multidimensional platform of patient-derived tumors identifies drug susceptibilities for clinical lenvatinib resistance.Acta Pharm Sin B. 2024 Jan;14(1):223-240. doi: 10.1016/j.apsb.2023.09.015. Epub 2023 Sep 25. Acta Pharm Sin B. 2024. PMID: 38261805 Free PMC article.
-
Exploring Tumor-Immune Interactions in Co-Culture Models of T Cells and Tumor Organoids Derived from Patients.Int J Mol Sci. 2023 Sep 27;24(19):14609. doi: 10.3390/ijms241914609. Int J Mol Sci. 2023. PMID: 37834057 Free PMC article. Review.
-
A pancreatic cancer organoid-in-matrix platform shows distinct sensitivities to T cell killing.Sci Rep. 2024 Apr 23;14(1):9377. doi: 10.1038/s41598-024-60107-5. Sci Rep. 2024. PMID: 38654067 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials