

HIV-1 protease inhibitors with a P1 phosphonate modification maintain potency against drug-resistant variants by increased interactions with flap residues
- PMID: 37244161
- PMCID: PMC10332405
- DOI: 10.1016/j.ejmech.2023.115501
HIV-1 protease inhibitors with a P1 phosphonate modification maintain potency against drug-resistant variants by increased interactions with flap residues
Abstract
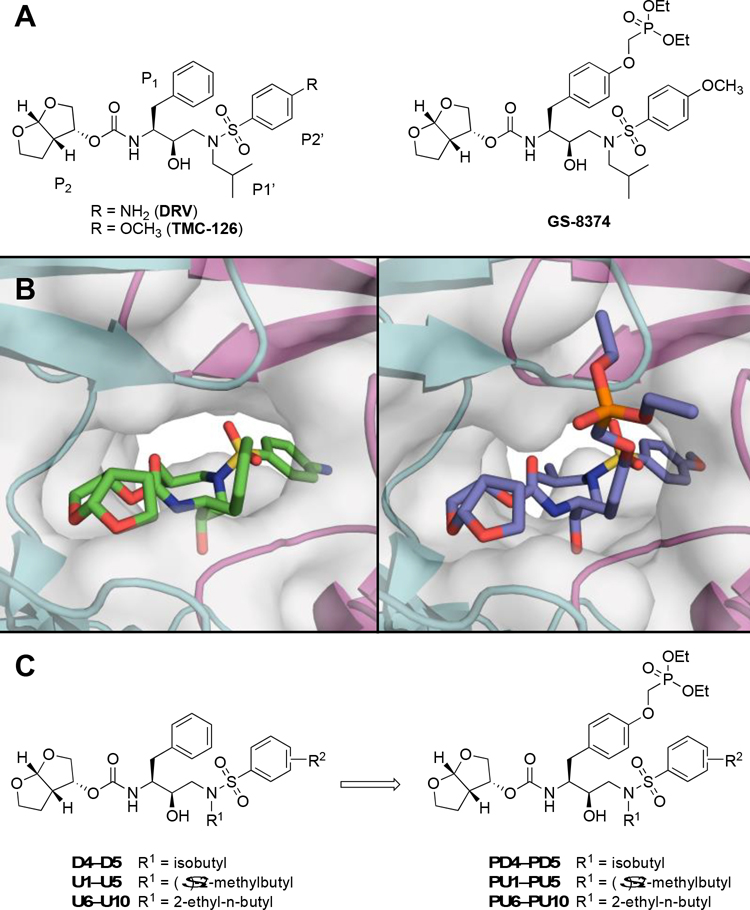
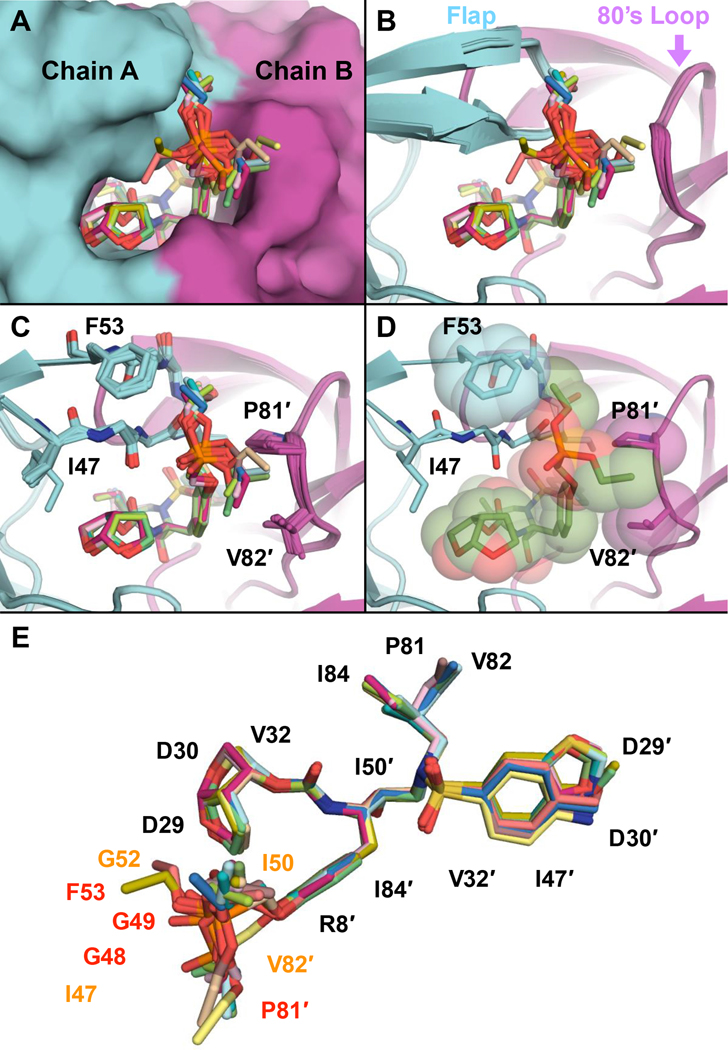
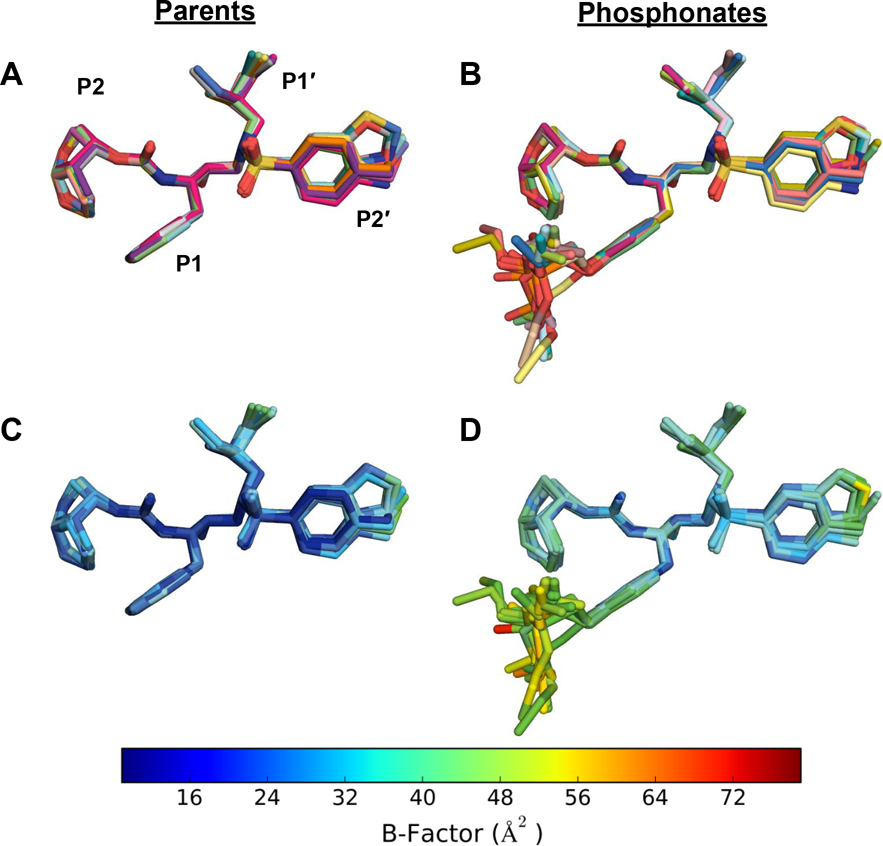
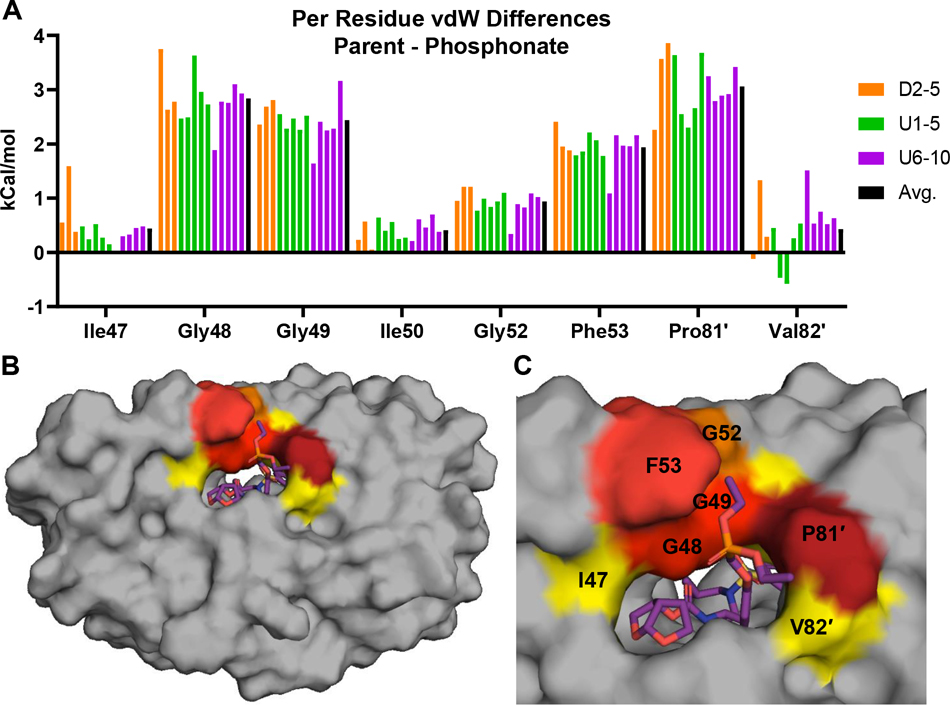
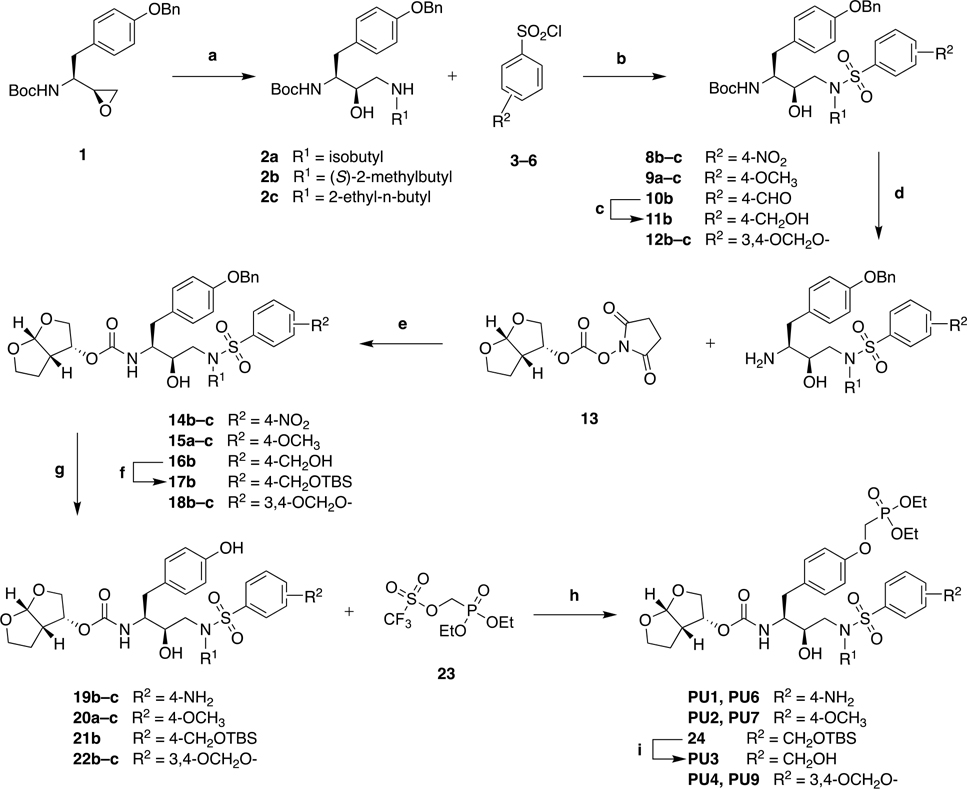
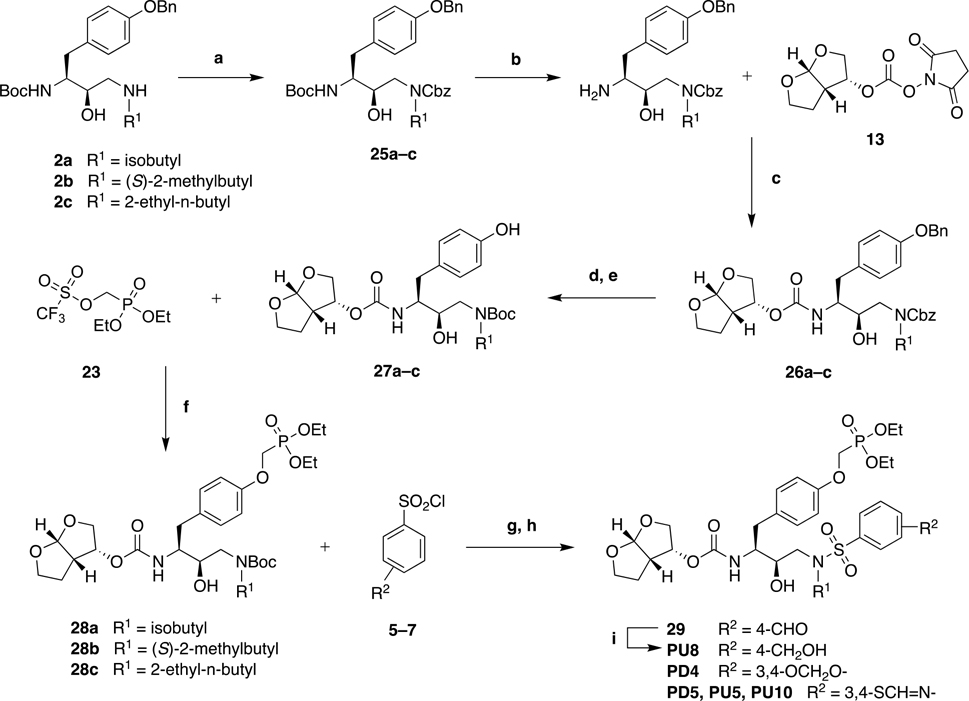
Protease inhibitors are the most potent antivirals against HIV-1, but they still lose efficacy against resistant variants. Improving the resistance profile is key to developing more robust inhibitors, which may be promising candidates for simplified next-generation antiretroviral therapies. In this study, we explored analogs of darunavir with a P1 phosphonate modification in combination with increasing size of the P1' hydrophobic group and various P2' moieties to improve potency against resistant variants. The phosphonate moiety substantially improved potency against highly mutated and resistant HIV-1 protease variants, but only when combined with more hydrophobic moieties at the P1' and P2' positions. Phosphonate analogs with a larger hydrophobic P1' moiety maintained excellent antiviral potency against a panel of highly resistant HIV-1 variants, with significantly improved resistance profiles. The cocrystal structures indicate that the phosphonate moiety makes extensive hydrophobic interactions with the protease, especially with the flap residues. Many residues involved in these protease-inhibitor interactions are conserved, enabling the inhibitors to maintain potency against highly resistant variants. These results highlight the need to balance inhibitor physicochemical properties by simultaneous modification of chemical groups to further improve resistance profiles.
Keywords: Drug resistance; HIV-1 protease; Protease inhibitors; SAR studies; X-ray structure.
Copyright © 2023 Elsevier Masson SAS. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
Structural Adaptation of Darunavir Analogues against Primary Mutations in HIV-1 Protease.ACS Infect Dis. 2019 Feb 8;5(2):316-325. doi: 10.1021/acsinfecdis.8b00336. Epub 2018 Dec 31. ACS Infect Dis. 2019. PMID: 30543749 Free PMC article.
-
GRL-079, a Novel HIV-1 Protease Inhibitor, Is Extremely Potent against Multidrug-Resistant HIV-1 Variants and Has a High Genetic Barrier against the Emergence of Resistant Variants.Antimicrob Agents Chemother. 2018 Apr 26;62(5):e02060-17. doi: 10.1128/AAC.02060-17. Print 2018 May. Antimicrob Agents Chemother. 2018. PMID: 29463535 Free PMC article.
-
A Modified P1 Moiety Enhances In Vitro Antiviral Activity against Various Multidrug-Resistant HIV-1 Variants and In Vitro Central Nervous System Penetration Properties of a Novel Nonpeptidic Protease Inhibitor, GRL-10413.Antimicrob Agents Chemother. 2016 Nov 21;60(12):7046-7059. doi: 10.1128/AAC.01428-16. Print 2016 Dec. Antimicrob Agents Chemother. 2016. PMID: 27620483 Free PMC article.
-
Beyond darunavir: recent development of next generation HIV-1 protease inhibitors to combat drug resistance.Chem Commun (Camb). 2022 Oct 20;58(84):11762-11782. doi: 10.1039/d2cc04541a. Chem Commun (Camb). 2022. PMID: 36200462 Free PMC article. Review.
-
Resilience to resistance of HIV-1 protease inhibitors: profile of darunavir.AIDS Rev. 2008 Jul-Sep;10(3):131-42. AIDS Rev. 2008. PMID: 18820715 Free PMC article. Review.
Cited by
-
Diastereoselective Synthesis of the HIV Protease Inhibitor Darunavir and Related Derivatives via a Titanium Tetrachloride-Mediated Asymmetric Glycolate Aldol Addition Reaction.J Org Chem. 2024 Jul 5;89(13):9569-9585. doi: 10.1021/acs.joc.4c01057. Epub 2024 Jun 25. J Org Chem. 2024. PMID: 38916048 Free PMC article.
-
Design, Synthesis, and Biological Evaluation of Darunavir Analogs as HIV-1 Protease Inhibitors.ACS Bio Med Chem Au. 2024 Sep 19;4(5):242-256. doi: 10.1021/acsbiomedchemau.4c00040. eCollection 2024 Oct 16. ACS Bio Med Chem Au. 2024. PMID: 39431267 Free PMC article.
-
FMO-guided design of darunavir analogs as HIV-1 protease inhibitors.Sci Rep. 2024 Feb 13;14(1):3639. doi: 10.1038/s41598-024-53940-1. Sci Rep. 2024. PMID: 38351065 Free PMC article.
References
-
- World Health Organization (WHO). HIV/AIDS, Fact Sheet (Updated November 2022). https://www.who.int/news-room/fact-sheets/detail/hiv-aids. 2022.
-
- Zhan P, Pannecouque C, De Clercq E, Liu X Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem, 59 (2016), pp. 2849–2878. - PubMed
-
- Gandhi RT, Bedimo R, Hoy JF, Landovitz RJ, Smith DM, Eaton EF, Lehmann C, Springer SA, Sax PE, Thompson MA, Benson CA, Buchbinder SP, Del Rio C, Eron JJ Jr., Gunthard HF, Molina JM, Jacobsen DM, Saag MS Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2022 recommendations of the International Antiviral Society-USA Panel. JAMA, 329 (2023), pp. 63–84. - PubMed
-
- Cihlar T, Fordyce M Current status and prospects of HIV treatment. Curr. Opin. Virol, 18 (2016), pp. 50–56. - PubMed
-
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV; Department of Health and Human Services. Available at https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv. (Accessed January 15, 2022).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
