

A New Generation of IMiDs as Treatments for Neuroinflammatory and Neurodegenerative Disorders
- PMID: 37238617
- PMCID: PMC10216254
- DOI: 10.3390/biom13050747
A New Generation of IMiDs as Treatments for Neuroinflammatory and Neurodegenerative Disorders
Abstract
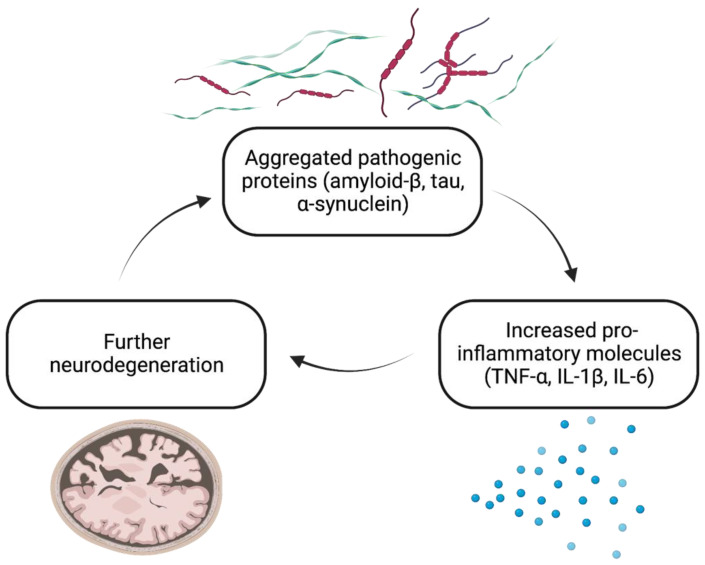
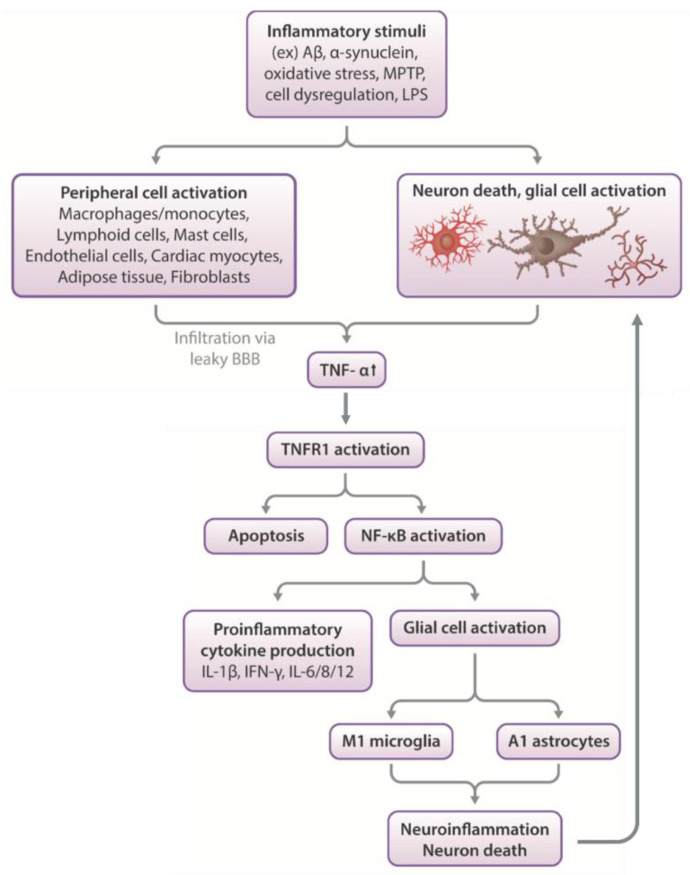
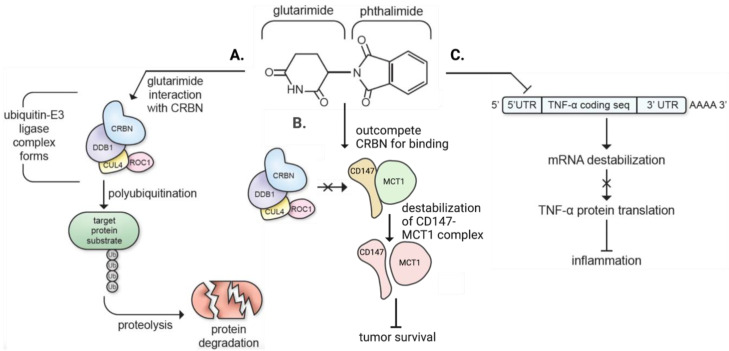
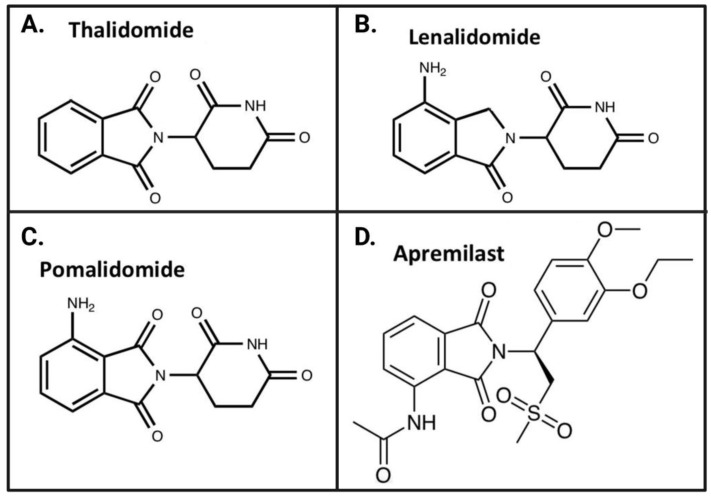
The immunomodulatory imide drug (IMiD) class, which includes the founding drug member thalidomide and later generation drugs, lenalidomide and pomalidomide, has dramatically improved the clinical treatment of specific cancers, such as multiple myeloma, and it combines potent anticancer and anti-inflammatory actions. These actions, in large part, are mediated by IMiD binding to the human protein cereblon that forms a critical component of the E3 ubiquitin ligase complex. This complex ubiquitinates and thereby regulates the levels of multiple endogenous proteins. However, IMiD-cereblon binding modifies cereblon's normal targeted protein degradation towards a new set of neosubstrates that underlies the favorable pharmacological action of classical IMiDs, but also their adverse actions-in particular, their teratogenicity. The ability of classical IMiDs to reduce the synthesis of key proinflammatory cytokines, especially TNF-α levels, makes them potentially valuable to reposition as drugs to mitigate inflammatory-associated conditions and, particularly, neurological disorders driven by an excessive neuroinflammatory element, as occurs in traumatic brain injury, Alzheimer's and Parkinson's diseases, and ischemic stroke. The teratogenic and anticancer actions of classical IMiDs are substantial liabilities for effective drugs in these disorders and can theoretically be dialed out of the drug class. We review a select series of novel IMiDs designed to avoid binding with human cereblon and/or evade degradation of downstream neosubstrates considered to underpin the adverse actions of thalidomide-like drugs. These novel non-classical IMiDs hold potential as new medications for erythema nodosum leprosum (ENL), a painful inflammatory skin condition associated with Hansen's disease for which thalidomide remains widely used, and, in particular, as a new treatment strategy for neurodegenerative disorders in which neuroinflammation is a key component.
Keywords: Alzheimer’s disease; Parkinson’s disease; cereblon; erythema nodosum leprosum; immunomodulatory imide drugs (IMiDs); neurodegeneration; neuroinflammation; pomalidomide; thalidomide; traumatic brain injury.
Conflict of interest statement
N.H.G., D.T., S-C.H., N.V., and D.S.K. are inventors on patents related to novel thalidomide and pomalidomide analogs. They have assigned their rights in entirety to their respective institutions, and hence have no personal rights to these patents. The National Institute on Aging, NIH, has a Cooperative Research and Development Agreement with AevisBio Inc. (Daejeon, Republic of Korea) to support the evaluation of novel IMiDs in neurological disorders. DSK is an employee and shareholder in AevisBio Inc (Daejeon, Republic of Korea). All other authors declare no conflict of interest.
Figures


Similar articles
-
Novel, thalidomide-like, non-cereblon binding drug tetrafluorobornylphthalimide mitigates inflammation and brain injury.J Biomed Sci. 2023 Mar 6;30(1):16. doi: 10.1186/s12929-023-00907-5. J Biomed Sci. 2023. PMID: 36872339 Free PMC article.
-
USP15 antagonizes CRL4CRBN-mediated ubiquitylation of glutamine synthetase and neosubstrates.Proc Natl Acad Sci U S A. 2021 Oct 5;118(40):e2111391118. doi: 10.1073/pnas.2111391118. Proc Natl Acad Sci U S A. 2021. PMID: 34583995 Free PMC article.
-
Generation of a lenalidomide-sensitive syngeneic murine in vivo multiple myeloma model by expression of CrbnI391V.Exp Hematol. 2021 Jan;93:61-69.e4. doi: 10.1016/j.exphem.2020.11.004. Epub 2020 Nov 11. Exp Hematol. 2021. PMID: 33186626
-
Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs.Int J Hematol. 2016 Sep;104(3):293-9. doi: 10.1007/s12185-016-2073-4. Epub 2016 Jul 26. Int J Hematol. 2016. PMID: 27460676 Review.
-
Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma.Leuk Lymphoma. 2013 Apr;54(4):683-7. doi: 10.3109/10428194.2012.728597. Epub 2012 Sep 28. Leuk Lymphoma. 2013. PMID: 22966948 Free PMC article. Review.
Cited by
-
Type 2 diabetes mellitus/obesity drugs: A neurodegenerative disorders savior or a bridge too far?Ageing Res Rev. 2024 Jul;98:102343. doi: 10.1016/j.arr.2024.102343. Epub 2024 May 16. Ageing Res Rev. 2024. PMID: 38762101 Review.
-
Neuroinflammation of Microglial Regulation in Alzheimer's Disease: Therapeutic Approaches.Molecules. 2024 Mar 26;29(7):1478. doi: 10.3390/molecules29071478. Molecules. 2024. PMID: 38611758 Free PMC article. Review.
-
Cancer drugs with high repositioning potential for Alzheimer's disease.Expert Opin Emerg Drugs. 2023 Dec;28(4):311-332. doi: 10.1080/14728214.2023.2296079. Epub 2023 Dec 26. Expert Opin Emerg Drugs. 2023. PMID: 38100555 Free PMC article. Review.
-
Neuroprotective Strategies and Cell-Based Biomarkers for Manganese-Induced Toxicity in Human Neuroblastoma (SH-SY5Y) Cells.Biomolecules. 2024 May 31;14(6):647. doi: 10.3390/biom14060647. Biomolecules. 2024. PMID: 38927051 Free PMC article.
-
Can Some Anticancer Drugs Be Repurposed to Treat Amyotrophic Lateral Sclerosis? A Brief Narrative Review.Int J Mol Sci. 2024 Feb 1;25(3):1751. doi: 10.3390/ijms25031751. Int J Mol Sci. 2024. PMID: 38339026 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
