

This is a preprint.
Human cytomegalovirus attenuates AKT activity by destabilizing insulin receptor substrate proteins
- PMID: 37131605
- PMCID: PMC10153195
- DOI: 10.1101/2023.04.17.537203
Human cytomegalovirus attenuates AKT activity by destabilizing insulin receptor substrate proteins
Update in
-
Human cytomegalovirus attenuates AKT activity by destabilizing insulin receptor substrate proteins.J Virol. 2023 Oct 31;97(10):e0056323. doi: 10.1128/jvi.00563-23. Epub 2023 Sep 27. J Virol. 2023. PMID: 37754763 Free PMC article.
Abstract
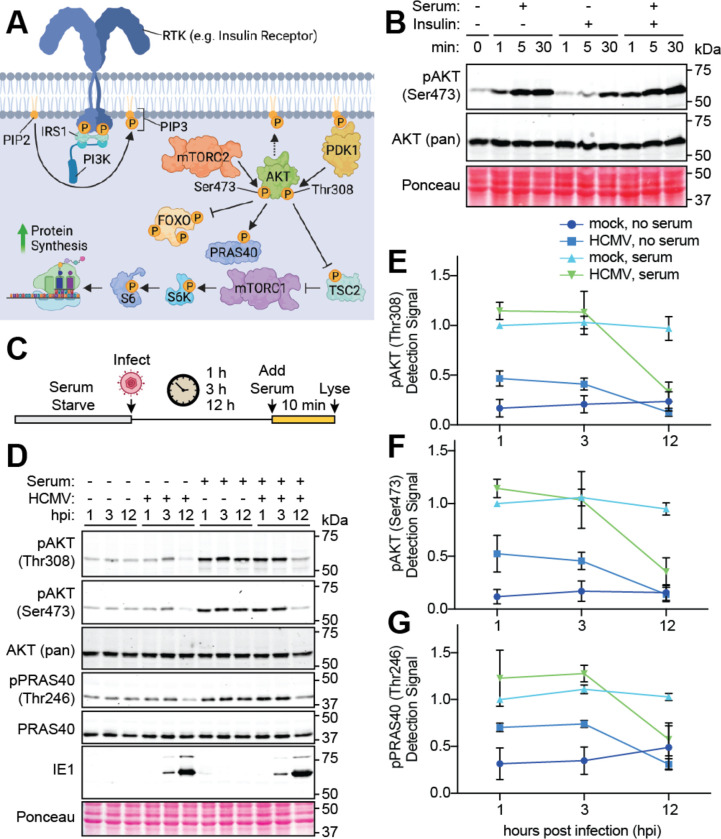
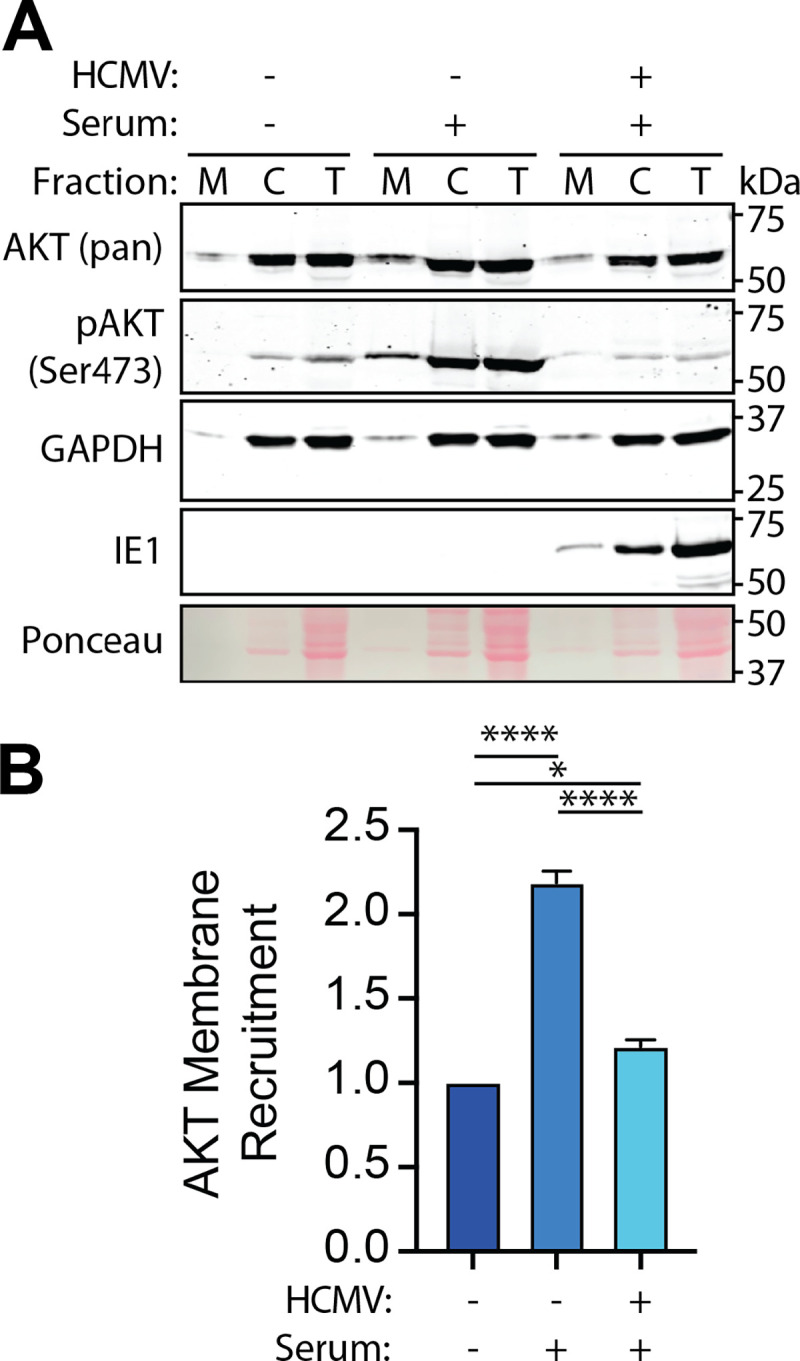
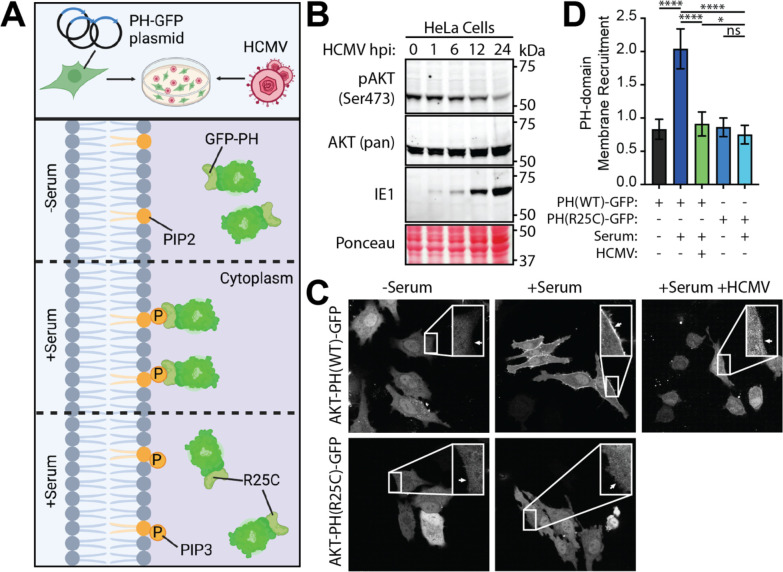
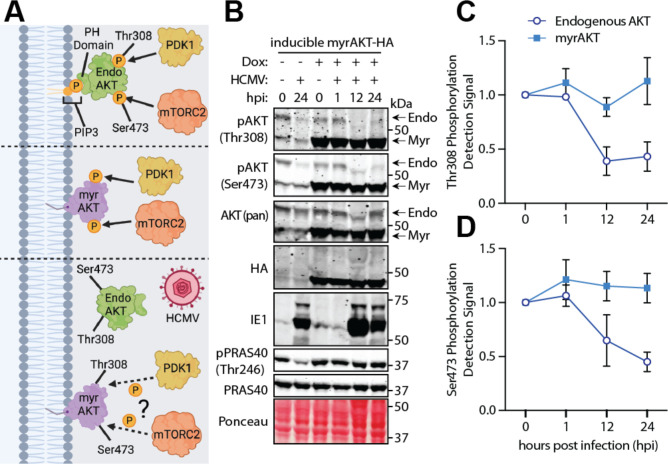
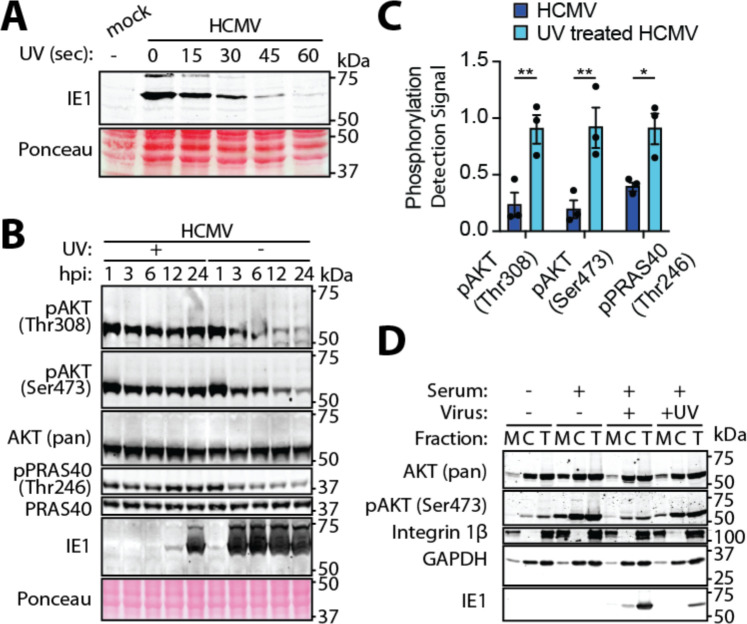
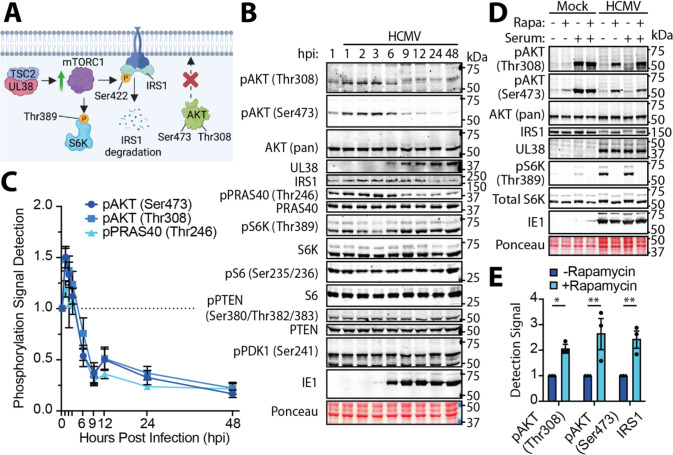
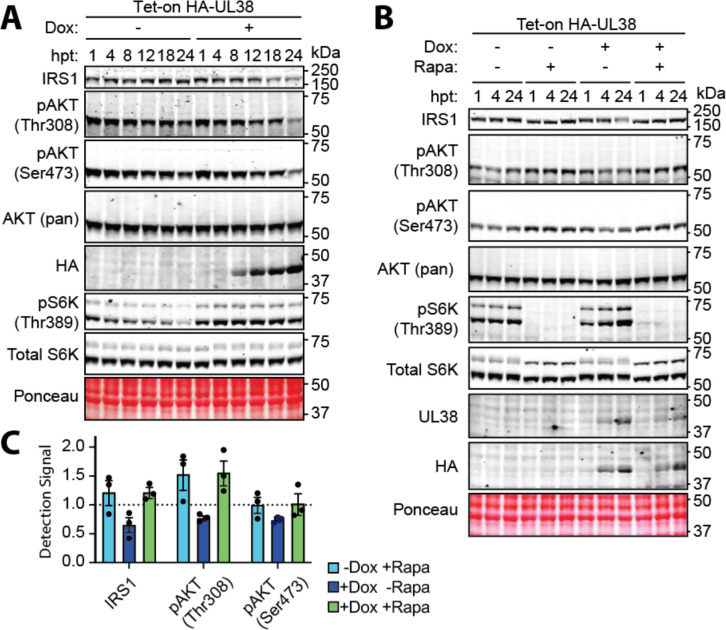
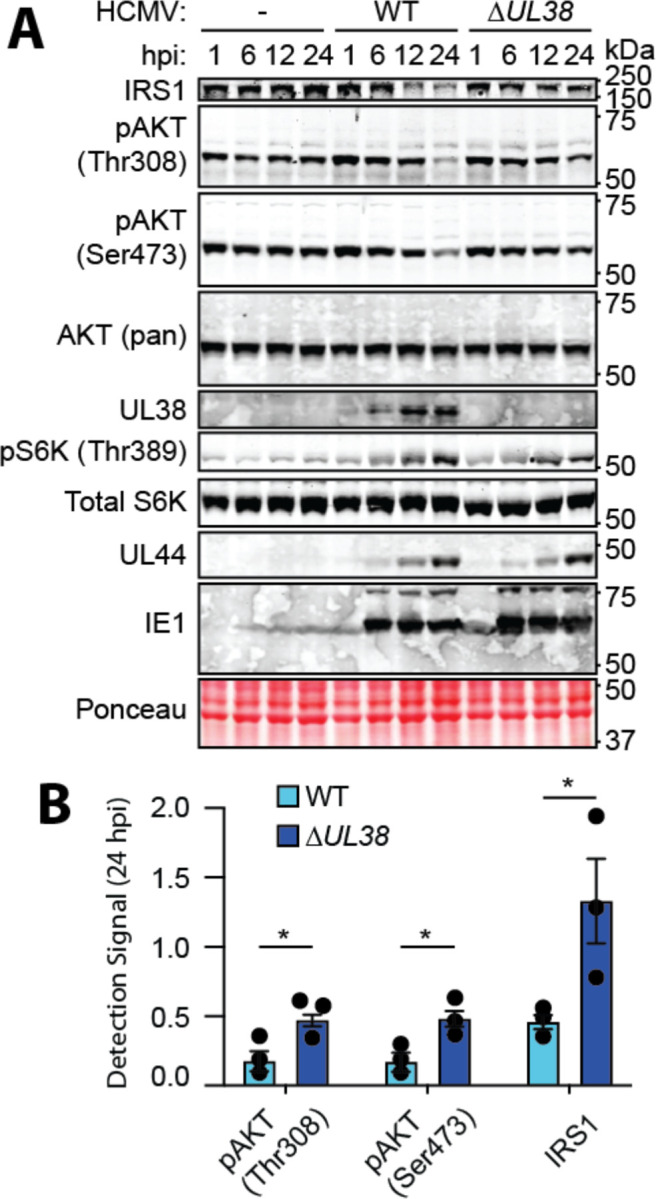
The phosphoinositide 3-kinase (PI3K)/AKT pathway plays crucial roles in cell viability and protein synthesis and is frequently co-opted by viruses to support their replication. Although many viruses maintain high levels of AKT activity during infection, other viruses, such as vesicular stomatitis virus and human cytomegalovirus (HCMV), cause AKT to accumulate in an inactive state. To efficiently replicate, HCMV requires FoxO transcription factors to localize to the infected cell nucleus (Zhang et. al. mBio 2022), a process directly antagonized by AKT. Therefore, we sought to investigate how HCMV inactivates AKT to achieve this. Subcellular fractionation and live cell imaging studies indicated that AKT failed to recruit to membranes upon serum-stimulation of infected cells. However, UV-inactivated virions were unable to render AKT non-responsive to serum, indicating a requirement for de novo viral gene expression. Interestingly, we were able to identify that UL38 (pUL38), a viral activator of mTORC1, is required to diminish AKT responsiveness to serum. mTORC1 contributes to insulin resistance by causing proteasomal degradation of insulin receptor substrate (IRS) proteins, such as IRS1, which are necessary for the recruitment of PI3K to growth factor receptors. In cells infected with a recombinant HCMV disrupted for UL38 , AKT responsiveness to serum is retained and IRS1 is not degraded. Furthermore, ectopic expression of UL38 in uninfected cells induces IRS1 degradation, inactivating AKT. These effects of UL38 were reversed by the mTORC1 inhibitor, rapamycin. Collectively, our results demonstrate that HCMV relies upon a cell-intrinsic negative feedback loop to render AKT inactive during productive infection.
Figures
Similar articles
-
Human cytomegalovirus attenuates AKT activity by destabilizing insulin receptor substrate proteins.J Virol. 2023 Oct 31;97(10):e0056323. doi: 10.1128/jvi.00563-23. Epub 2023 Sep 27. J Virol. 2023. PMID: 37754763 Free PMC article.
-
The Akt Forkhead Box O Transcription Factor Axis Regulates Human Cytomegalovirus Replication.mBio. 2022 Aug 30;13(4):e0104222. doi: 10.1128/mbio.01042-22. Epub 2022 Aug 10. mBio. 2022. PMID: 35946797 Free PMC article.
-
Tuberous Sclerosis Complex Protein 2-Independent Activation of mTORC1 by Human Cytomegalovirus pUL38.J Virol. 2015 Aug;89(15):7625-35. doi: 10.1128/JVI.01027-15. Epub 2015 May 13. J Virol. 2015. PMID: 25972538 Free PMC article.
-
[Interrelationship between human cytomegalovirus infection and chemokine].Nihon Rinsho. 1998 Jan;56(1):69-74. Nihon Rinsho. 1998. PMID: 9465667 Review. Japanese.
-
New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia.Cell Signal. 2014 Jan;26(1):149-61. doi: 10.1016/j.cellsig.2013.09.021. Epub 2013 Oct 16. Cell Signal. 2014. PMID: 24140475 Review.
References
-
- Xiang K, Wang B. Role of the PI3K-AKT-mTOR pathway in hepatitis B virus infection and replication. Mol Med Rep. 2018;17: 4713–4719. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials