

Connecting the dots: Neuronal senescence, stress granules, and neurodegeneration
- PMID: 37084987
- PMCID: PMC10205695
- DOI: 10.1016/j.gene.2023.147437
Connecting the dots: Neuronal senescence, stress granules, and neurodegeneration
Abstract
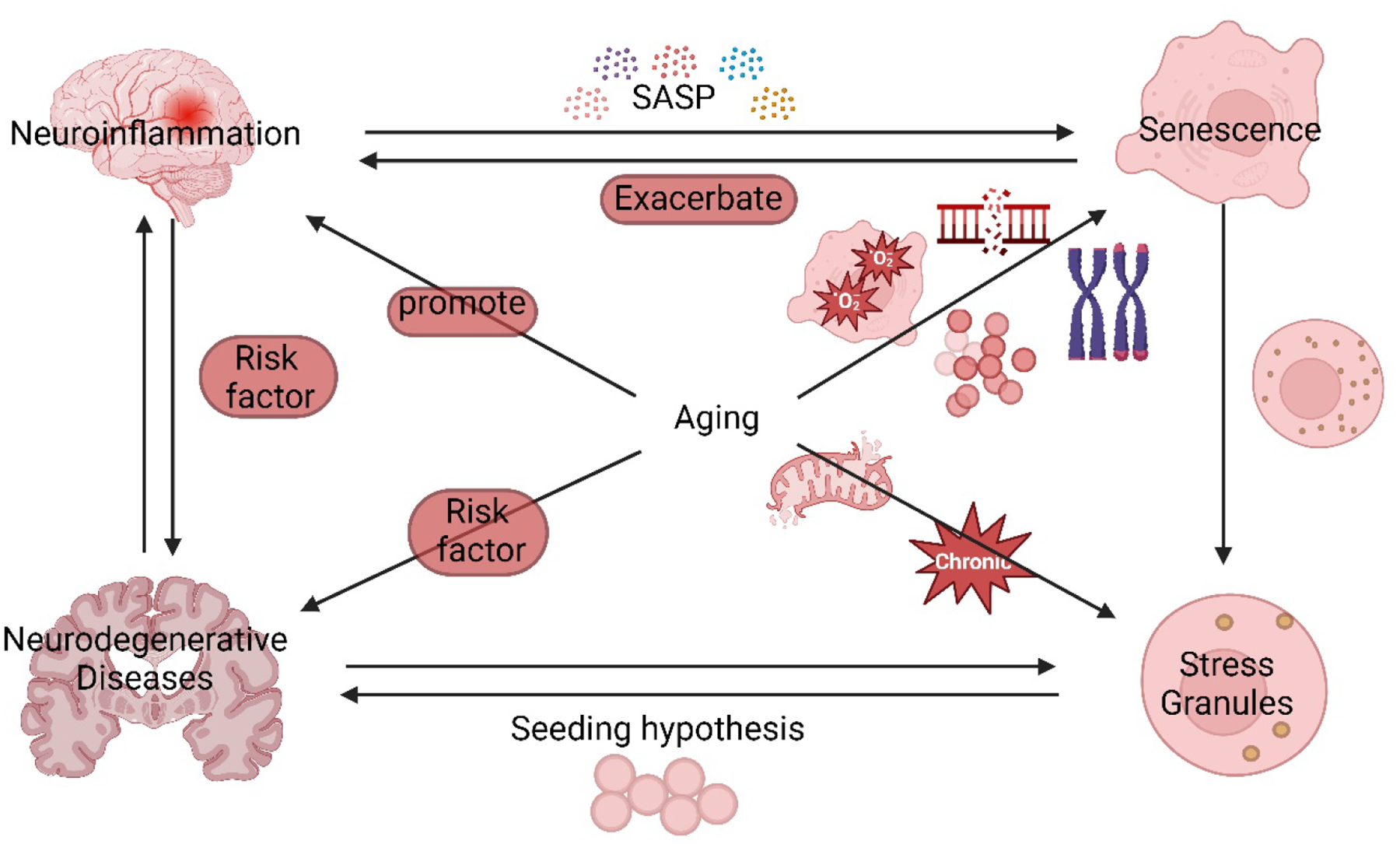
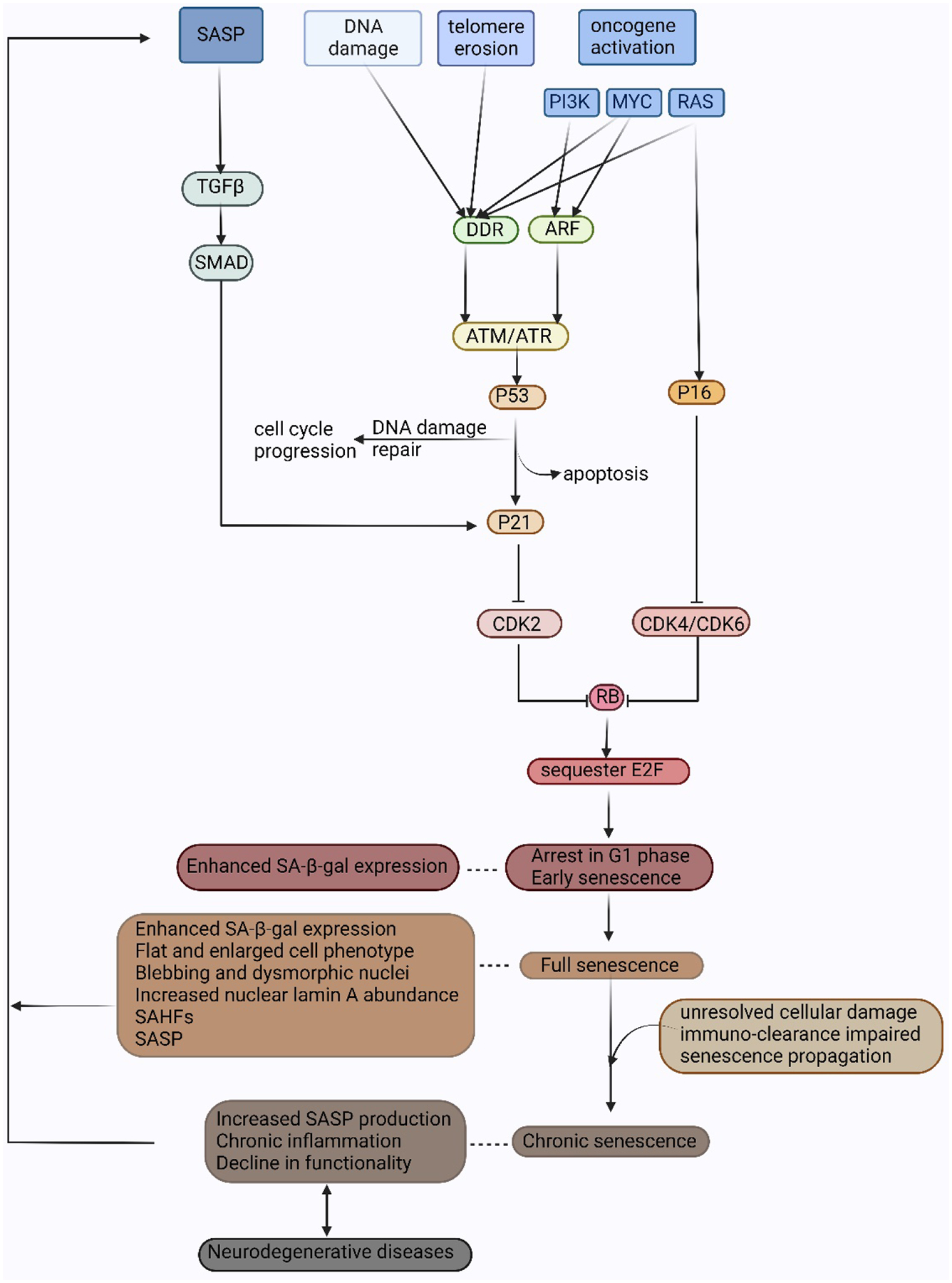
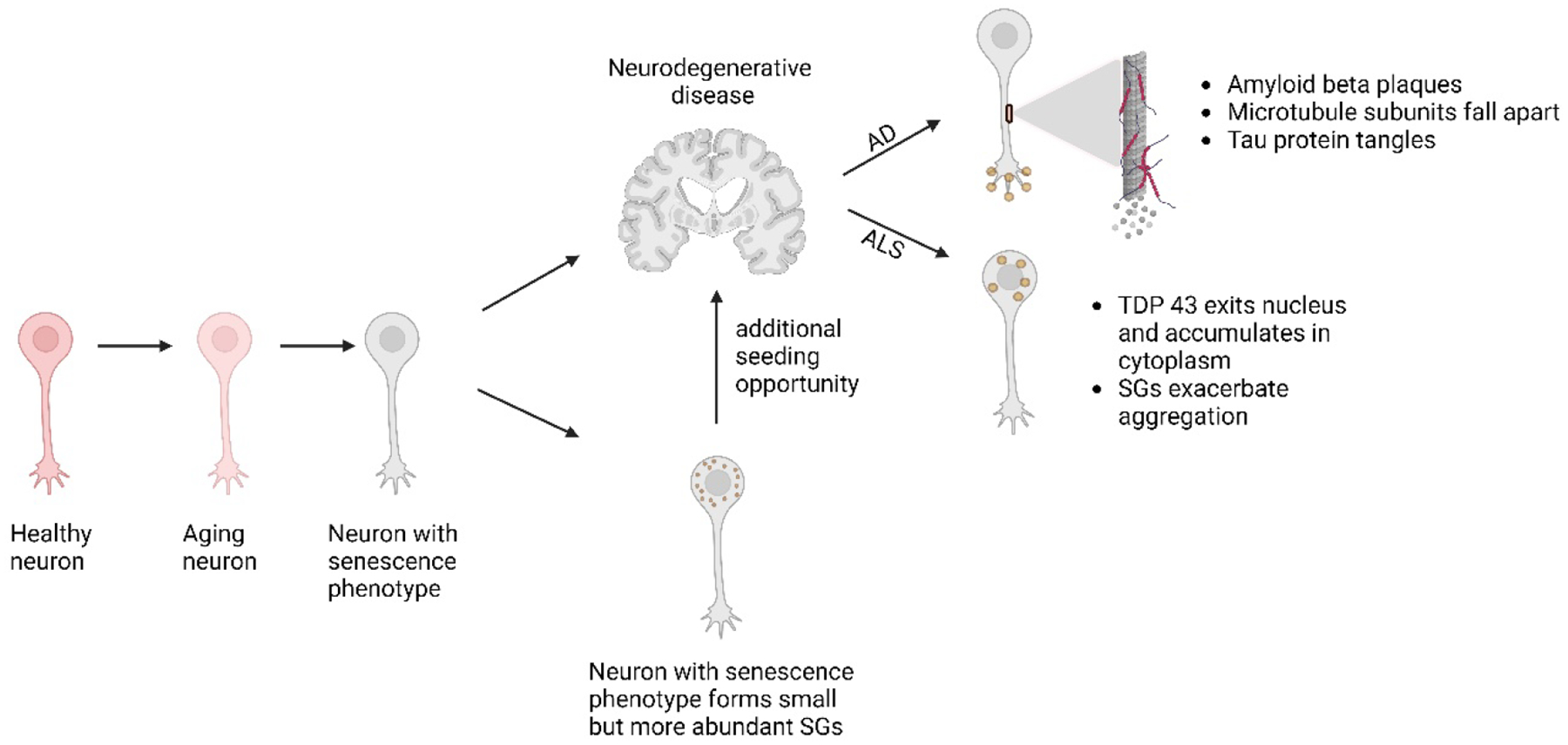
Cellular senescence increases with aging. While senescence is associated with an exit of the cell cycle, there is ample evidence that post-mitotic cells including neurons can undergo senescence as the brain ages, and that senescence likely contributes significantly to the progression of neurodegenerative diseases (ND) such as Alzheimer's Disease (AD) and Amyotrophic Lateral Sclerosis (ALS). Stress granules (SGs) are stress-induced cytoplasmic biomolecular condensates of RNA and proteins, which have been linked to the development of AD and ALS. The SG seeding hypothesis of NDs proposes that chronic stress in aging neurons results in static SGs that progress into pathological aggregates Alterations in SG dynamics have also been linked to senescence, though studies that link SGs and senescence in the context of NDs and the aging brain have not yet been performed. In this Review, we summarize the literature on senescence, and explore the contribution of senescence to the aging brain. We describe senescence phenotypes in aging neurons and glia, and their links to neuroinflammation and the development of AD and ALS. We further examine the relationships of SGs to senescence and to ND. We propose a new hypothesis that neuronal senescence may contribute to the mechanism of SG seeding in ND by altering SG dynamics in aged cells, thereby providing additional aggregation opportunities within aged neurons.
Keywords: Alzheimer’s Disease; Amyotrophic Lateral Sclerosis (ALS); Neurodegeneration; Neuroinflammation; Neuronal aging; Senescence; Senescence-associated secretory phenotype (SASP); Stress granules.
Copyright © 2023 Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
Targeting stress granules in neurodegenerative diseases: A focus on biological function and dynamics disorders.Biofactors. 2024 May-Jun;50(3):422-438. doi: 10.1002/biof.2017. Epub 2023 Nov 15. Biofactors. 2024. PMID: 37966813 Review.
-
Stress granules in neurodegeneration--lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma.FEBS J. 2013 Sep;280(18):4348-70. doi: 10.1111/febs.12287. Epub 2013 May 9. FEBS J. 2013. PMID: 23587065 Review.
-
Stress Granule Homeostasis, Aberrant Phase Transition, and Amyotrophic Lateral Sclerosis.ACS Chem Neurosci. 2022 Aug 17;13(16):2356-2370. doi: 10.1021/acschemneuro.2c00262. Epub 2022 Jul 29. ACS Chem Neurosci. 2022. PMID: 35905138 Review.
-
Molecular interaction of stress granules with Tau and autophagy in Alzheimer's disease.Neurochem Int. 2022 Jul;157:105342. doi: 10.1016/j.neuint.2022.105342. Epub 2022 Apr 21. Neurochem Int. 2022. PMID: 35461975 Review.
-
Stress granule-mediated RNA regulatory mechanism in Alzheimer's disease.Geriatr Gerontol Int. 2024 Mar;24 Suppl 1:7-14. doi: 10.1111/ggi.14663. Epub 2023 Sep 19. Geriatr Gerontol Int. 2024. PMID: 37726158 Review.
Cited by
-
Alzheimer's Disease and Green Tea: Epigallocatechin-3-Gallate as a Modulator of Inflammation and Oxidative Stress.Antioxidants (Basel). 2023 Jul 20;12(7):1460. doi: 10.3390/antiox12071460. Antioxidants (Basel). 2023. PMID: 37507998 Free PMC article. Review.
-
Molecular hallmarks of ageing in amyotrophic lateral sclerosis.Cell Mol Life Sci. 2024 Mar 2;81(1):111. doi: 10.1007/s00018-024-05164-9. Cell Mol Life Sci. 2024. PMID: 38430277 Free PMC article. Review.
-
Endogenous TDP-43 mislocalization in a novel knock-in mouse model reveals DNA repair impairment, inflammation, and neuronal senescence.Res Sq [Preprint]. 2024 Mar 20:rs.3.rs-3879966. doi: 10.21203/rs.3.rs-3879966/v1. Res Sq. 2024. PMID: 38343852 Free PMC article. Preprint.
References
-
- Al-Rushadi M, Musabah Rashid. (2022). Investigation of the role of G3BP in stress granule formation in senescence. Doctoral thesis, Durham University. Available from: http://etheses.dur.ac.uk/14347/
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
