

Genetic variability in sporadic amyotrophic lateral sclerosis
- PMID: 37043475
- PMCID: PMC10473563
- DOI: 10.1093/brain/awad120
Genetic variability in sporadic amyotrophic lateral sclerosis
Abstract
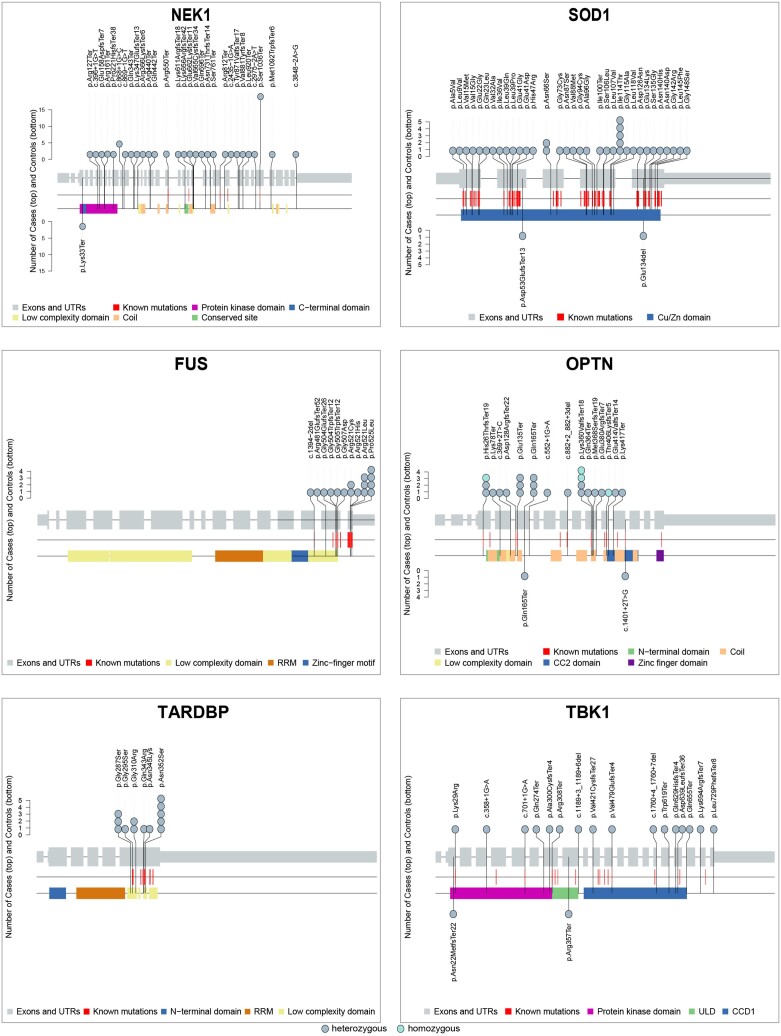
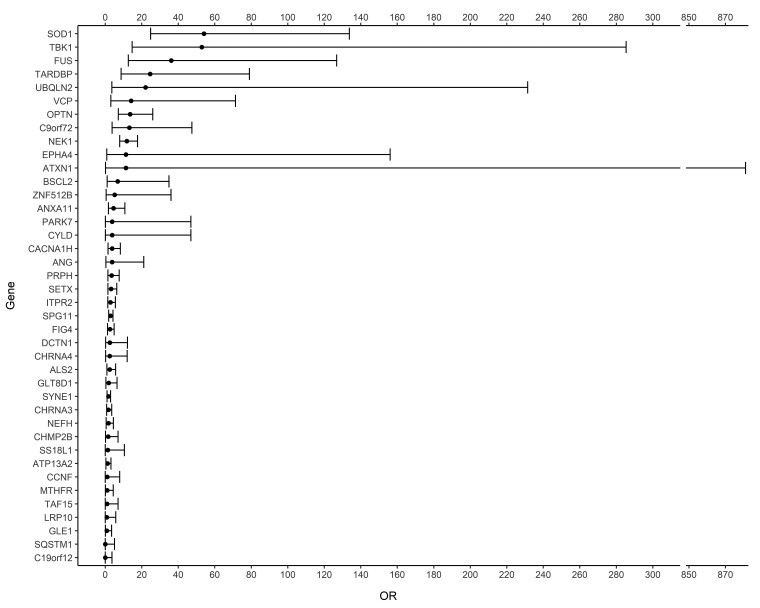
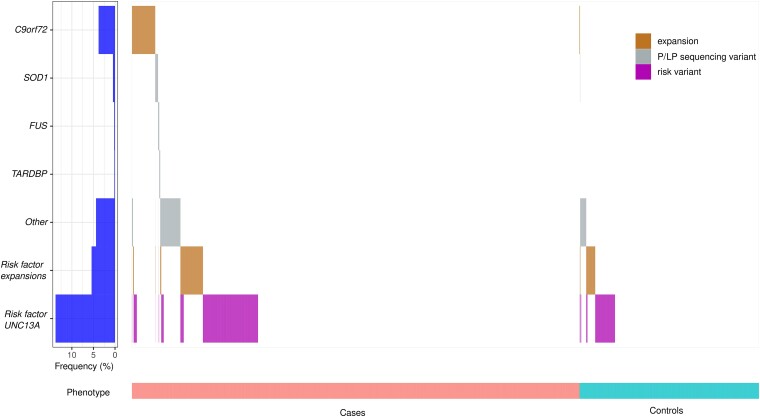
With the advent of gene therapies for amyotrophic lateral sclerosis (ALS), there is a surge in gene testing for this disease. Although there is ample experience with gene testing for C9orf72, SOD1, FUS and TARDBP in familial ALS, large studies exploring genetic variation in all ALS-associated genes in sporadic ALS (sALS) are still scarce. Gene testing in a diagnostic setting is challenging, given the complex genetic architecture of sALS, for which there are genetic variants with large and small effect sizes. Guidelines for the interpretation of genetic variants in gene panels and for counselling of patients are lacking. We aimed to provide a thorough characterization of genetic variability in ALS genes by applying the American College of Medical Genetics and Genomics (ACMG) criteria on whole genome sequencing data from a large cohort of 6013 sporadic ALS patients and 2411 matched controls from Project MinE. We studied genetic variation in 90 ALS-associated genes and applied customized ACMG-criteria to identify pathogenic and likely pathogenic variants. Variants of unknown significance were collected as well. In addition, we determined the length of repeat expansions in C9orf72, ATXN1, ATXN2 and NIPA1 using the ExpansionHunter tool. We found C9orf72 repeat expansions in 5.21% of sALS patients. In 50 ALS-associated genes, we did not identify any pathogenic or likely pathogenic variants. In 5.89%, a pathogenic or likely pathogenic variant was found, most commonly in SOD1, TARDBP, FUS, NEK1, OPTN or TBK1. Significantly more cases carried at least one pathogenic or likely pathogenic variant compared to controls (odds ratio 1.75; P-value 1.64 × 10-5). Isolated risk factors in ATXN1, ATXN2, NIPA1 and/or UNC13A were detected in 17.33% of cases. In 71.83%, we did not find any genetic clues. A combination of variants was found in 2.88%. This study provides an inventory of pathogenic and likely pathogenic genetic variation in a large cohort of sALS patients. Overall, we identified pathogenic and likely pathogenic variants in 11.13% of ALS patients in 38 known ALS genes. In line with the oligogenic hypothesis, we found significantly more combinations of variants in cases compared to controls. Many variants of unknown significance may contribute to ALS risk, but diagnostic algorithms to reliably identify and weigh them are lacking. This work can serve as a resource for counselling and for the assembly of gene panels for ALS. Further characterization of the genetic architecture of sALS is necessary given the growing interest in gene testing in ALS.
Keywords: complex genetic disease; motor neuron disease; oligogenic inheritance.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
J.V. reports to have sponsored research agreements with Biogen and Astra Zeneca. A.A.-C reports consultancies or advisory boards for Amylyx, Apellis, Biogen, Brainstorm, Cytokinetics, GenieUs, GSK, Lilly, Mitsubishi Tanabe Pharma, Novartis, OrionPharma, Quralis, Sano, Sanofi and Wave Pharmaceuticals. P.V.D. has served in advisory boards for Biogen, CSL Behring, Alexion Pharmaceuticals, Ferrer, QurAlis, Cytokinetics, Argenx, UCB, Muna Therapeutics, Alector, Augustine Therapeutics, VectorY and Zambon.
Figures
Similar articles
-
Genetics screening in an Italian cohort of patients with Amyotrophic Lateral Sclerosis: the importance of early testing and its implication.J Neurol. 2024 Apr;271(4):1921-1936. doi: 10.1007/s00415-023-12142-x. Epub 2023 Dec 19. J Neurol. 2024. PMID: 38112783
-
Role of genetics in amyotrophic lateral sclerosis: a large cohort study in Chinese mainland population.J Med Genet. 2022 Sep;59(9):840-849. doi: 10.1136/jmedgenet-2021-107965. Epub 2021 Sep 20. J Med Genet. 2022. PMID: 34544842 Free PMC article.
-
Genotype-phenotype correlation in Tunisian patients with Amyotrophic Lateral Sclerosis.Neurobiol Aging. 2022 Dec;120:27-33. doi: 10.1016/j.neurobiolaging.2022.08.002. Epub 2022 Aug 14. Neurobiol Aging. 2022. PMID: 36108486
-
Estimated Prevalence and Incidence of Amyotrophic Lateral Sclerosis and SOD1 and C9orf72 Genetic Variants.Neuroepidemiology. 2021;55(5):342-353. doi: 10.1159/000516752. Epub 2021 Jul 9. Neuroepidemiology. 2021. PMID: 34247168 Review.
-
Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis.J Neurol Neurosurg Psychiatry. 2017 Jul;88(7):540-549. doi: 10.1136/jnnp-2016-315018. Epub 2017 Jan 5. J Neurol Neurosurg Psychiatry. 2017. PMID: 28057713 Review.
Cited by
-
RNA editing regulates glutamatergic synapses in the frontal cortex of a molecular subtype of Amyotrophic Lateral Sclerosis.Mol Med. 2024 Jul 12;30(1):101. doi: 10.1186/s10020-024-00863-2. Mol Med. 2024. PMID: 38997636 Free PMC article.
-
Molecular Mechanisms in the Design of Novel Targeted Therapies for Neurodegenerative Diseases.Curr Issues Mol Biol. 2024 May 29;46(6):5436-5453. doi: 10.3390/cimb46060325. Curr Issues Mol Biol. 2024. PMID: 38920997 Free PMC article. Review.
-
Early life events may be the first steps on the multistep path to amyotrophic lateral sclerosis.Sci Rep. 2024 Nov 18;14(1):28497. doi: 10.1038/s41598-024-78240-6. Sci Rep. 2024. PMID: 39557859 Free PMC article.
-
The genetics of amyotrophic lateral sclerosis.Curr Opin Neurol. 2024 Oct 1;37(5):560-569. doi: 10.1097/WCO.0000000000001294. Epub 2024 Aug 22. Curr Opin Neurol. 2024. PMID: 38967083 Free PMC article. Review.
-
NEK1 haploinsufficiency worsens DNA damage, but not defective ciliogenesis, in C9ORF72 patient-derived iPSC-motoneurons.Hum Mol Genet. 2024 Nov 5;33(21):1900-1907. doi: 10.1093/hmg/ddae121. Hum Mol Genet. 2024. PMID: 39222049 Free PMC article.
References
-
- Hardiman O, Al-Chalabi A, Chio A, et al. . Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17085. - PubMed
-
- Byrne S, Heverin M, Elamin M, et al. . Aggregation of neurologic and neuropsychiatric disease in amyotrophic lateral sclerosis kindreds: a population-based case-control cohort study of familial and sporadic amyotrophic lateral sclerosis. Ann Neurol. 2013;74:699–708. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
