

Lymphoma cells lacking pro-apoptotic BAX are highly resistant to BH3-mimetics targeting pro-survival MCL-1 but retain sensitivity to conventional DNA-damaging drugs
- PMID: 36755070
- PMCID: PMC10070326
- DOI: 10.1038/s41418-023-01117-0
Lymphoma cells lacking pro-apoptotic BAX are highly resistant to BH3-mimetics targeting pro-survival MCL-1 but retain sensitivity to conventional DNA-damaging drugs
Abstract
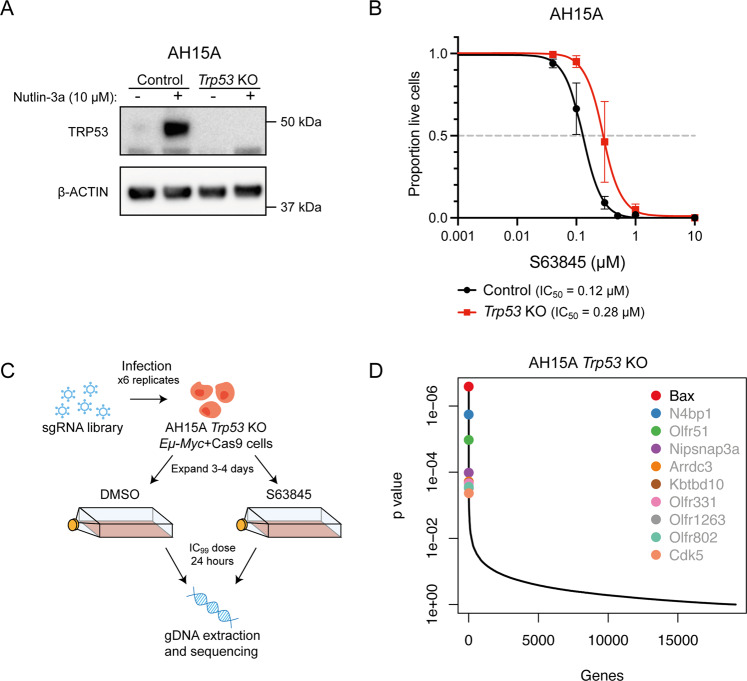
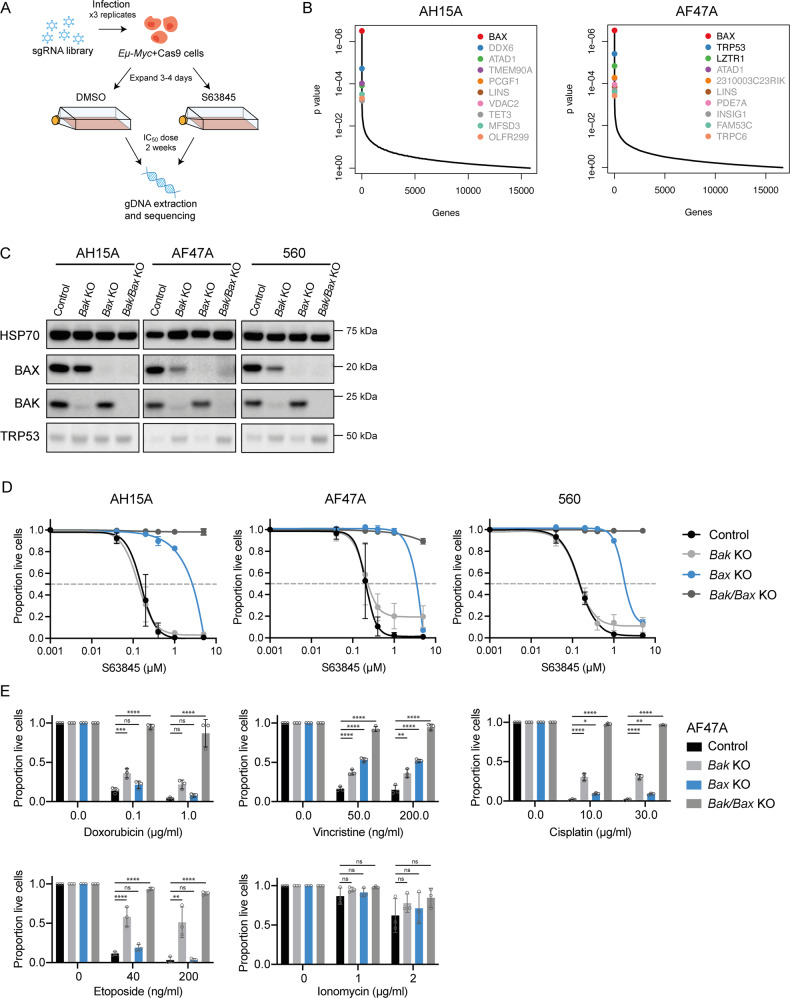
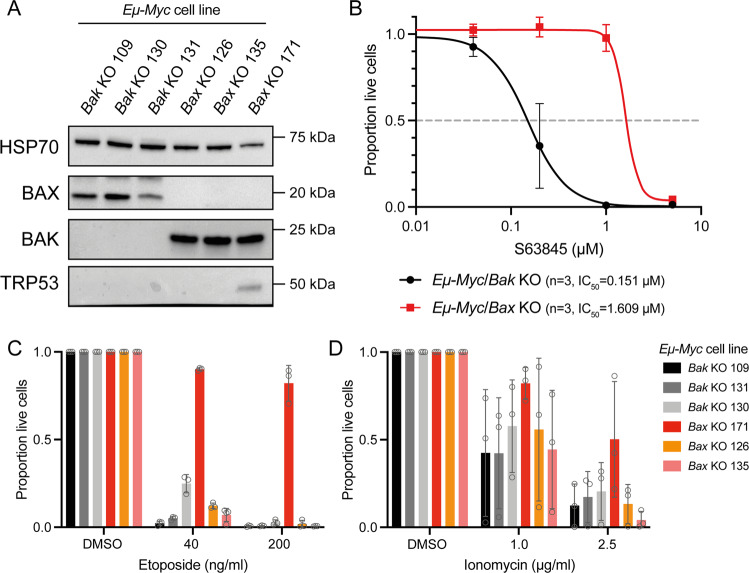
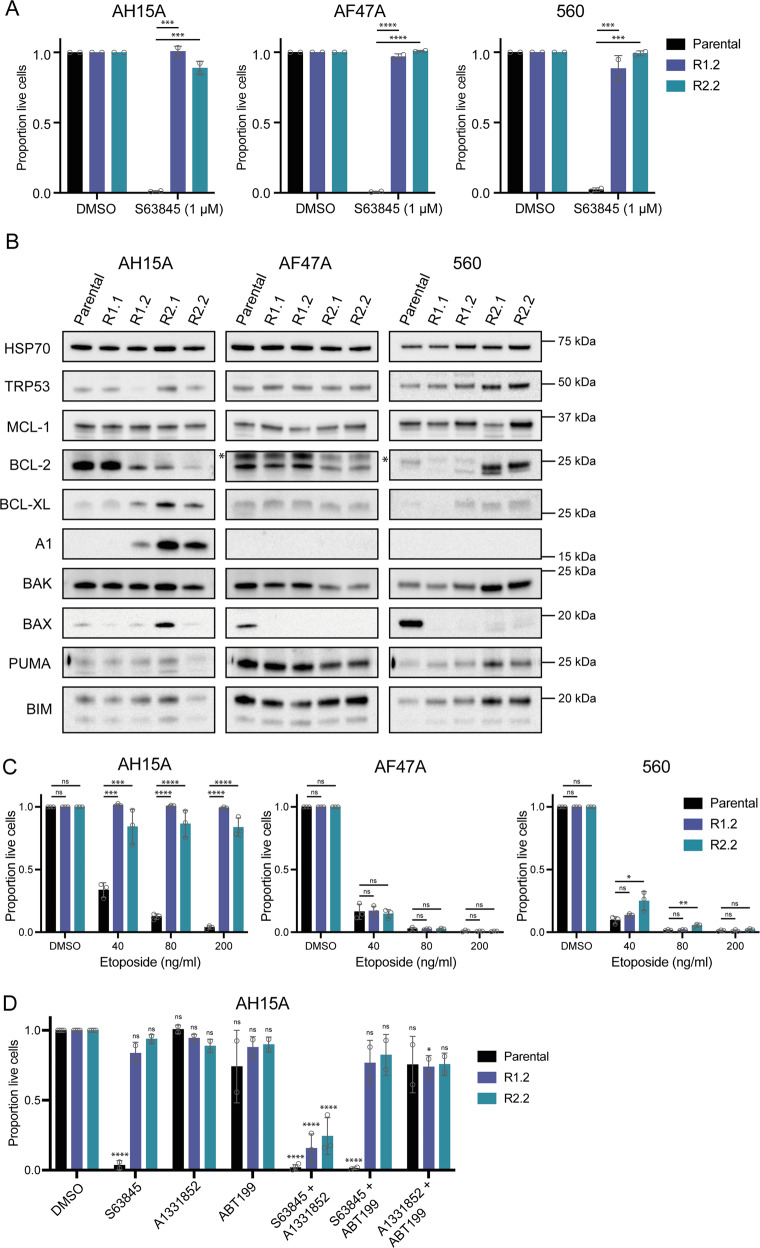
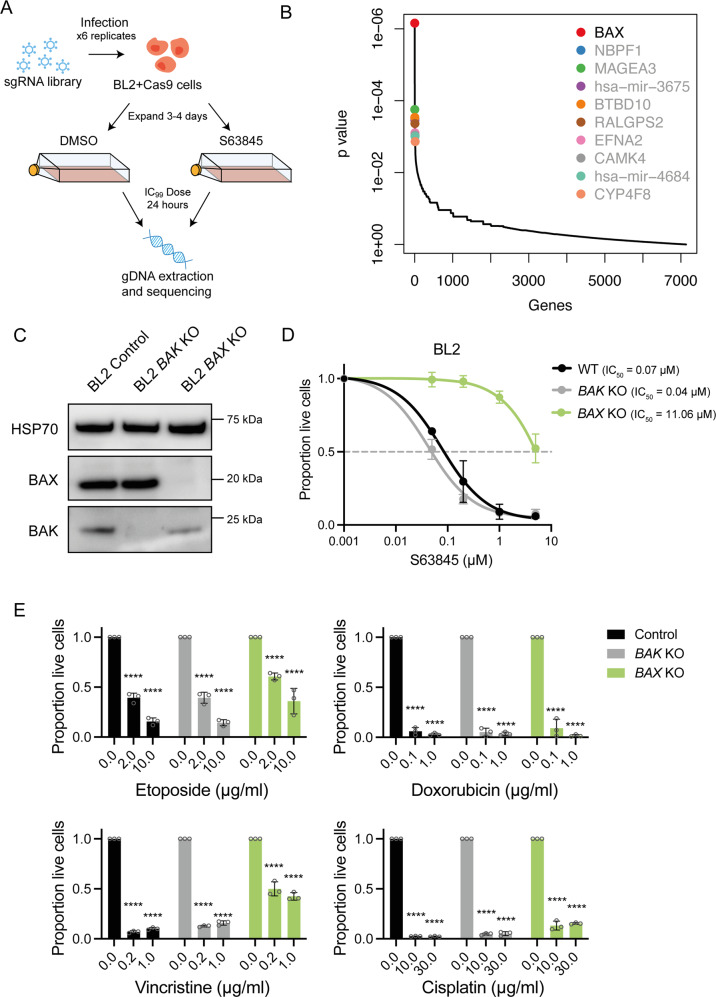
BH3-mimetic drugs are an anti-cancer therapy that can induce apoptosis in malignant cells by directly binding and inhibiting pro-survival proteins of the BCL-2 family. The BH3-mimetic drug venetoclax, which targets BCL-2, has been approved for the treatment of chronic lymphocytic leukaemia and acute myeloid leukaemia by regulatory authorities worldwide. However, while most patients initially respond well, resistance and relapse while on this drug is an emerging and critical issue in the clinic. Though some studies have begun uncovering the factors involved in resistance to BCL-2-targeting BH3-mimetic drugs, little focus has been applied to pre-emptively tackle resistance for the next generation of BH3-mimetic drugs targeting MCL-1, which are now in clinical trials for diverse blood cancers. Therefore, using pre-clinical mouse and human models of aggressive lymphoma, we sought to predict factors likely to contribute to the development of resistance in patients receiving MCL-1-targeting BH3-mimetic drugs. First, we performed multiple whole genome CRISPR/Cas9 KO screens and identified that loss of the pro-apoptotic effector protein BAX, but not its close relative BAK, could confer resistance to MCL-1-targeting BH3-mimetic drugs in both short-term and long-term treatment regimens, even in lymphoma cells lacking the tumour suppressor TRP53. Furthermore, we found that mouse Eµ-Myc lymphoma cells selected for loss of BAX, as well as upregulation of the untargeted pro-survival BCL-2 family proteins BCL-XL and A1, when made naturally resistant to MCL-1 inhibitors by culturing them in increasing doses of drug over time, a situation mimicking the clinical application of these drugs. Finally, we identified therapeutic approaches which could overcome these two methods of resistance: the use of chemotherapeutic drugs or combined BH3-mimetic treatment, respectively. Collectively, these results uncover some key factors likely to cause resistance to MCL-1 inhibition in the clinic and suggest rational therapeutic strategies to overcome resistance that should be investigated further.
© 2023. The Author(s).
Conflict of interest statement
All authors are employees of WEHI which receives milestone and royalty payments related to venetoclax. AS and GLK have received research funding from Servier.
Figures

Similar articles
-
The BH3 alpha-helical mimic BH3-M6 disrupts Bcl-X(L), Bcl-2, and MCL-1 protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a Bax- and Bim-dependent manner.J Biol Chem. 2011 Mar 18;286(11):9382-92. doi: 10.1074/jbc.M110.203638. Epub 2010 Dec 9. J Biol Chem. 2011. PMID: 21148306 Free PMC article.
-
BCL-W makes only minor contributions to MYC-driven lymphoma development.Oncogene. 2023 Sep;42(37):2776-2781. doi: 10.1038/s41388-023-02804-5. Epub 2023 Aug 11. Oncogene. 2023. PMID: 37567974 Free PMC article.
-
Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias.Blood. 2021 May 20;137(20):2721-2735. doi: 10.1182/blood.2020010167. Blood. 2021. PMID: 33824975 Free PMC article.
-
What can we learn from mice lacking pro-survival BCL-2 proteins to advance BH3 mimetic drugs for cancer therapy?Cell Death Differ. 2022 Jun;29(6):1079-1093. doi: 10.1038/s41418-022-00987-0. Epub 2022 Apr 6. Cell Death Differ. 2022. PMID: 35388168 Free PMC article. Review.
-
BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines.Cancer Cell. 2018 Dec 10;34(6):879-891. doi: 10.1016/j.ccell.2018.11.004. Cancer Cell. 2018. PMID: 30537511 Review.
Cited by
-
In situ visualization of endothelial cell-derived extracellular vesicle formation in steady state and malignant conditions.Nat Commun. 2024 Oct 22;15(1):8802. doi: 10.1038/s41467-024-52867-5. Nat Commun. 2024. PMID: 39438460 Free PMC article.
-
The Pivotal Role of Preclinical Animal Models in Anti-Cancer Drug Discovery and Personalized Cancer Therapy Strategies.Pharmaceuticals (Basel). 2024 Aug 9;17(8):1048. doi: 10.3390/ph17081048. Pharmaceuticals (Basel). 2024. PMID: 39204153 Free PMC article. Review.
-
Venetoclax resistance in acute lymphoblastic leukemia is characterized by increased mitochondrial activity and can be overcome by co-targeting oxidative phosphorylation.Cell Death Dis. 2024 Jul 3;15(7):475. doi: 10.1038/s41419-024-06864-7. Cell Death Dis. 2024. PMID: 38961053 Free PMC article.
-
Repurposing of Antidiarrheal Loperamide for Treating Melanoma by Inducing Cell Apoptosis and Cell Metastasis Suppression In vitro and In vivo.Curr Cancer Drug Targets. 2024;24(10):1015-1030. doi: 10.2174/0115680096283086240116093400. Curr Cancer Drug Targets. 2024. PMID: 38303527
-
Protective effects of dietary dimethyl itaconate supplementation on oxidative stress, inflammation, and apoptosis in broilers under chronic heat stress.J Anim Sci. 2023 Jan 3;101:skad356. doi: 10.1093/jas/skad356. J Anim Sci. 2023. PMID: 37837639 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
