

Investigational treatments for neurodegenerative diseases caused by inheritance of gene mutations: lessons from recent clinical trials
- PMID: 36751779
- PMCID: PMC10154469
- DOI: 10.4103/1673-5374.363185
Investigational treatments for neurodegenerative diseases caused by inheritance of gene mutations: lessons from recent clinical trials
Abstract
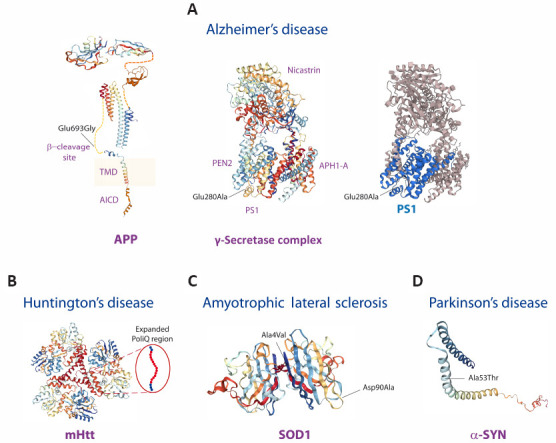
We reviewed recent major clinical trials with investigational drugs for the treatment of subjects with neurodegenerative diseases caused by inheritance of gene mutations or associated with genetic risk factors. Specifically, we discussed randomized clinical trials in subjects with Alzheimer's disease, Huntington's disease and amyotrophic lateral sclerosis bearing pathogenic gene mutations, and glucocerebrosidase-associated Parkinson's disease. Learning potential lessons to improve future therapeutic approaches is the aim of this review. Two long-term, controlled trials on three anti-β-amyloid monoclonal antibodies (solanezumab, gantenerumab and crenezumab) in subjects carrying Alzheimer's disease-linked mutated genes encoding for amyloid precursor protein or presenilin 1 or presenilin 2 failed to show cognitive or functional benefits. A major trial on tominersen, an antisense oligonucleotide designed to reduce the production of the huntingtin protein in subjects with Huntington's disease, was prematurely interrupted because the drug failed to show higher efficacy than placebo and, at highest doses, led to worsened outcomes. A 28-week trial of tofersen, an antisense oligonucleotide for superoxide dismutase 1 in patients with amyotrophic lateral sclerosis with superoxide dismutase 1 gene mutations failed to show significant beneficial effects but the 1-year open label extension of this study indicated better clinical and functional outcomes in the group with early tofersen therapy. A trial of venglustat, a potent and brain-penetrant glucosylceramide synthase inhibitor, in Parkinson's disease subjects with heterozygous glucocerebrosidase gene mutations revealed worsened clinical and cognitive performance of patients on the enzyme inhibitor compared to placebo. We concluded that clinical trials in neurodegenerative diseases with a genetic basis should test monoclonal antibodies, antisense oligonucleotides or gene editing directed against the mutated enzyme or the mutated substrate without dramatically affecting physiological wild-type variants.
Keywords: Alzheimer’s disease; Huntington’s disease; Parkinson’s disease; amyloid precursor protein; amyotrophic lateral sclerosis; glucocerebrosidase; huntingtin; presenilin 1; presenilin 2; superoxide dismutase 1.
Conflict of interest statement
None
Figures
Similar articles
-
Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases.Brain Res Bull. 2013 Aug;97:69-80. doi: 10.1016/j.brainresbull.2013.06.001. Epub 2013 Jun 10. Brain Res Bull. 2013. PMID: 23756188 Review.
-
Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS.N Engl J Med. 2022 Sep 22;387(12):1099-1110. doi: 10.1056/NEJMoa2204705. N Engl J Med. 2022. PMID: 36129998 Clinical Trial.
-
Protein misfolding in the late-onset neurodegenerative diseases: common themes and the unique case of amyotrophic lateral sclerosis.Proteins. 2013 Aug;81(8):1285-303. doi: 10.1002/prot.24285. Epub 2013 Jul 2. Proteins. 2013. PMID: 23508986 Review.
-
Antisense Drugs Make Sense for Neurological Diseases.Annu Rev Pharmacol Toxicol. 2021 Jan 6;61:831-852. doi: 10.1146/annurev-pharmtox-010919-023738. Epub 2020 Oct 9. Annu Rev Pharmacol Toxicol. 2021. PMID: 33035446 Free PMC article. Review.
-
Downregulation of glial genes involved in synaptic function mitigates Huntington's disease pathogenesis.Elife. 2021 Apr 19;10:e64564. doi: 10.7554/eLife.64564. Elife. 2021. PMID: 33871358 Free PMC article.
Cited by
-
Mechanism Exploration of Amyloid-β-42 Disaggregation by Single-Chain Variable Fragments of Alzheimer's Disease Therapeutic Antibodies.Int J Mol Sci. 2023 May 6;24(9):8371. doi: 10.3390/ijms24098371. Int J Mol Sci. 2023. PMID: 37176076 Free PMC article.
-
Looking to the Future: Drug Delivery and Targeting in the Prophylaxis and Therapy of Severe and Chronic Diseases.Handb Exp Pharmacol. 2024;284:389-411. doi: 10.1007/164_2023_696. Handb Exp Pharmacol. 2024. PMID: 37861719
-
Nucleic acid therapeutics as differentiation agents for myeloid leukemias.Leukemia. 2024 Jul;38(7):1441-1454. doi: 10.1038/s41375-024-02191-0. Epub 2024 Feb 29. Leukemia. 2024. PMID: 38424137 Free PMC article. Review.
-
Presenilin: A Multi-Functional Molecule in the Pathogenesis of Alzheimer's Disease and Other Neurodegenerative Diseases.Int J Mol Sci. 2024 Feb 1;25(3):1757. doi: 10.3390/ijms25031757. Int J Mol Sci. 2024. PMID: 38339035 Free PMC article. Review.
References
-
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ. Huntington disease. Nat Rev Dis Primers. (2015);1:15005. - PubMed
-
- Braak H, Del Tredici K, R¨ub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003);24:197–211. - PubMed
-
- Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. (2017);377:162–172. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
