

Synaptotagmin 9 Modulates Spontaneous Neurotransmitter Release in Striatal Neurons by Regulating Substance P Secretion
- PMID: 36732068
- PMCID: PMC9992334
- DOI: 10.1523/JNEUROSCI.1857-22.2023
Synaptotagmin 9 Modulates Spontaneous Neurotransmitter Release in Striatal Neurons by Regulating Substance P Secretion
Abstract
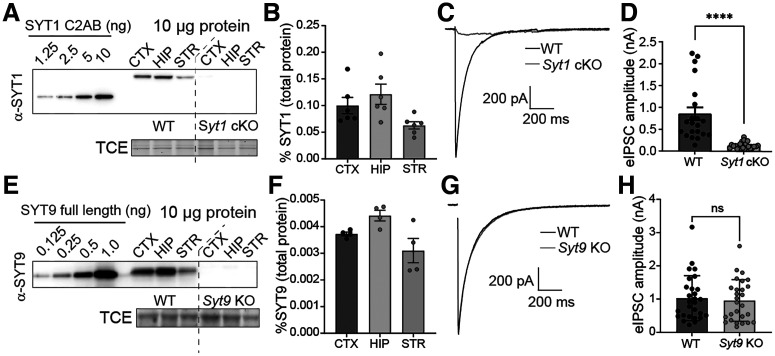
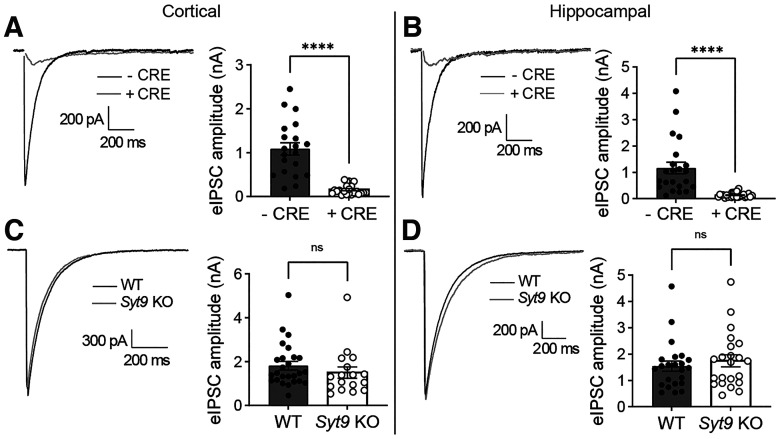
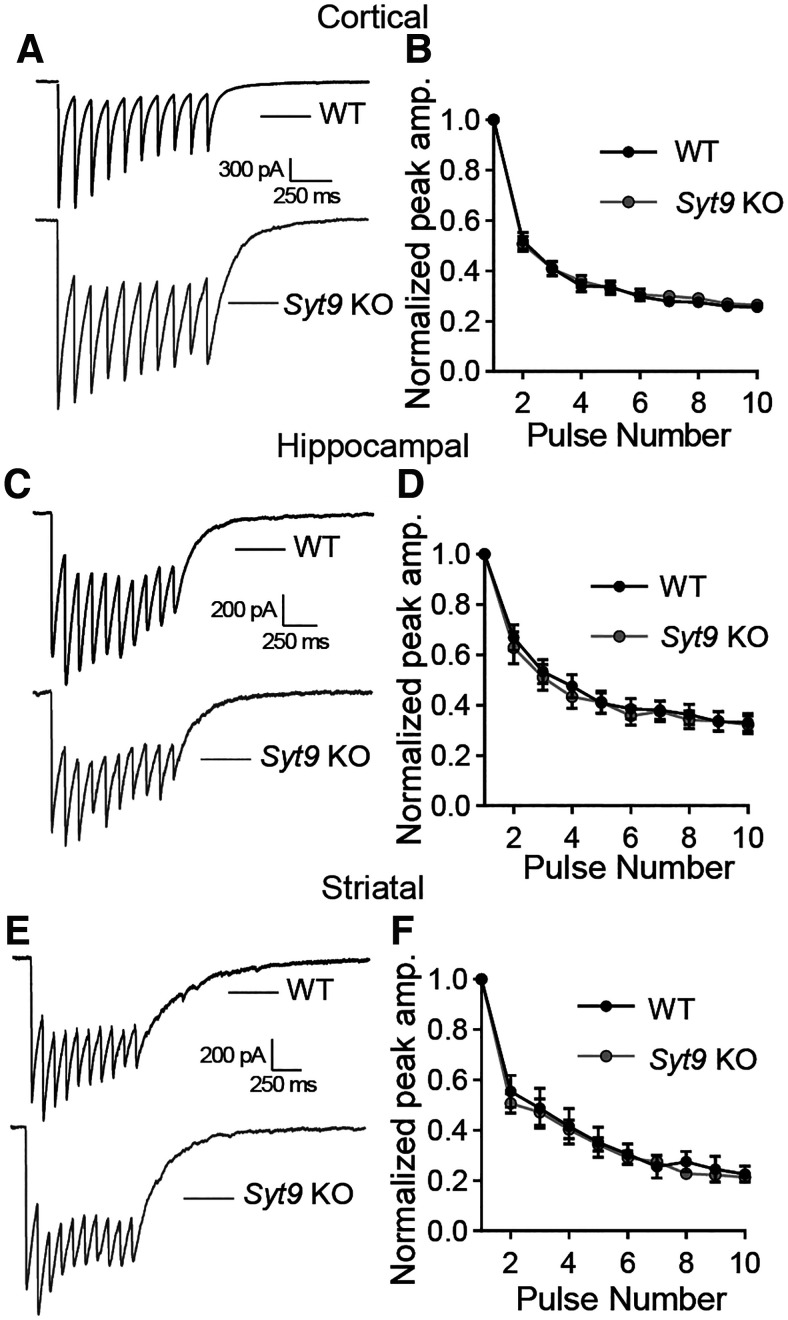
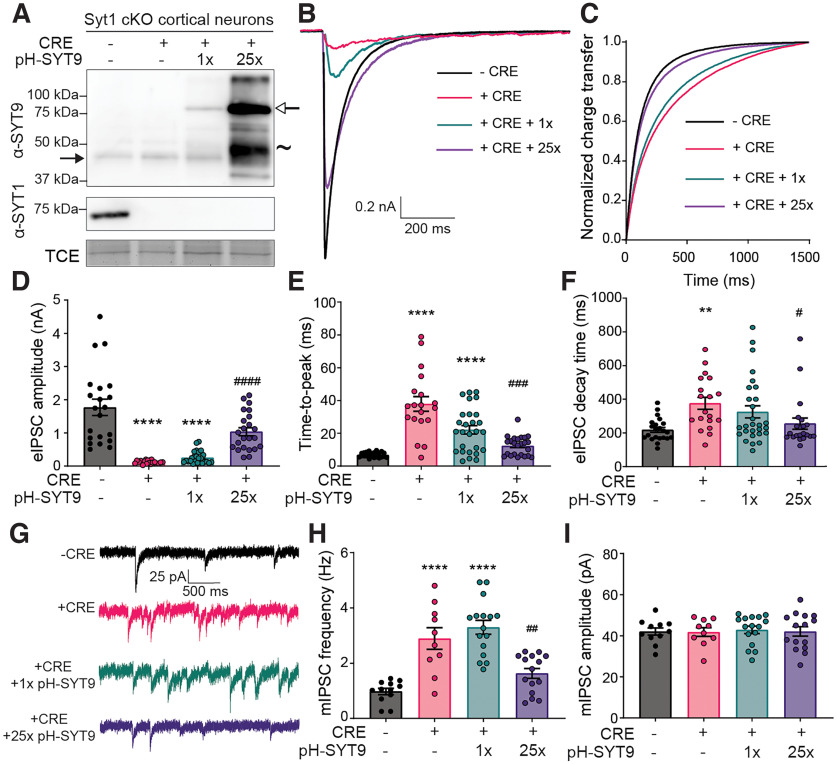
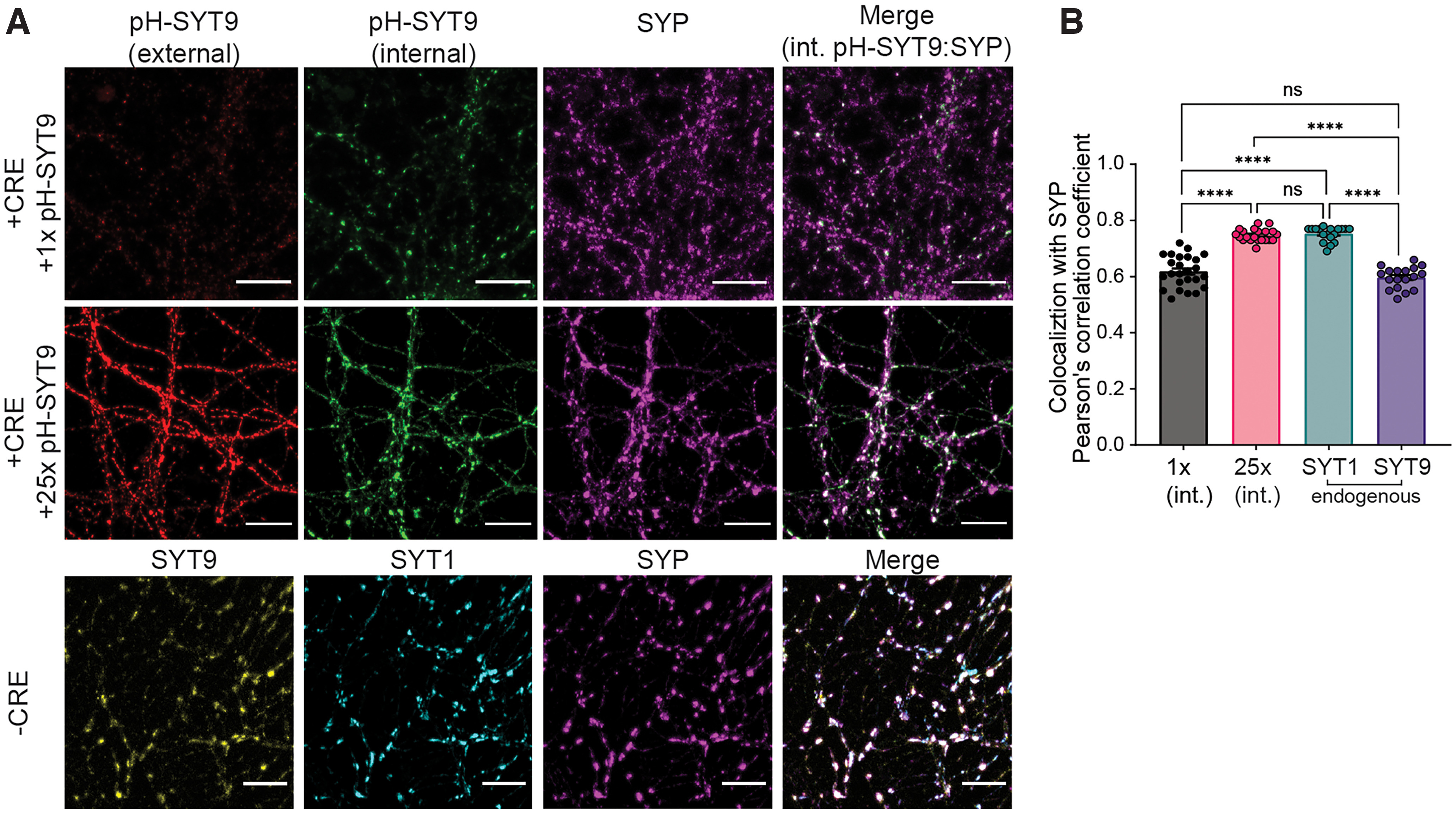
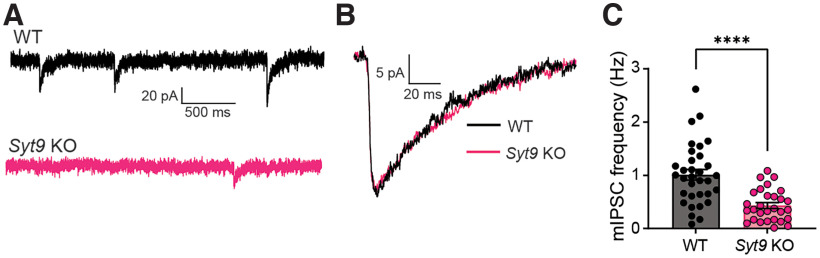
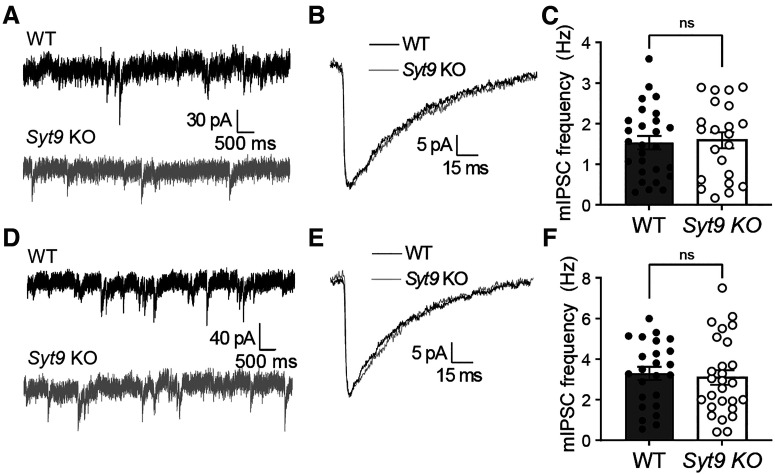
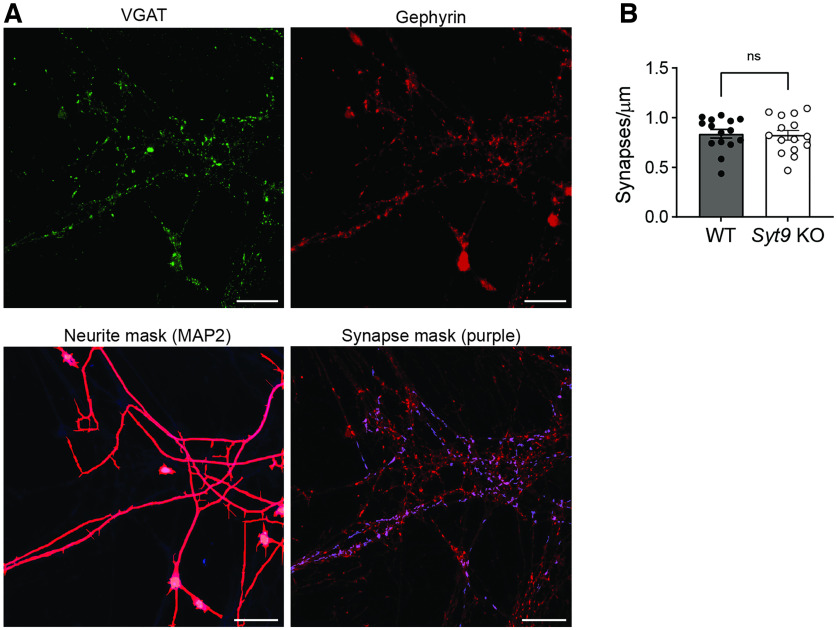
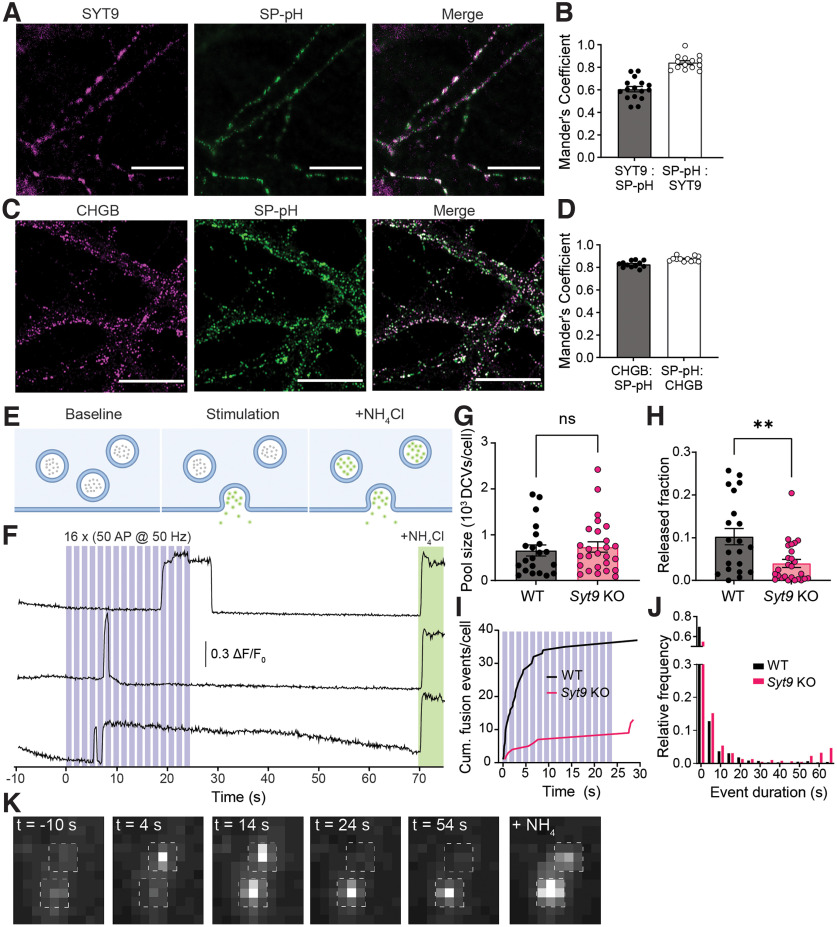
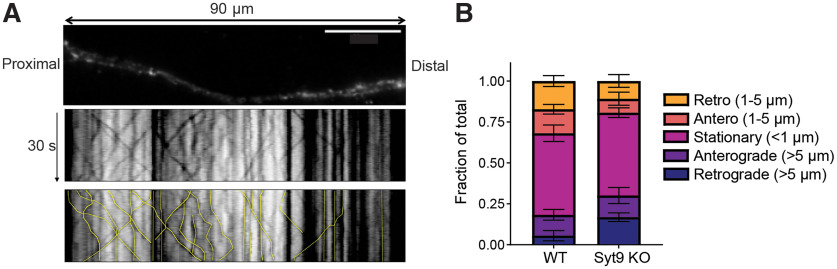
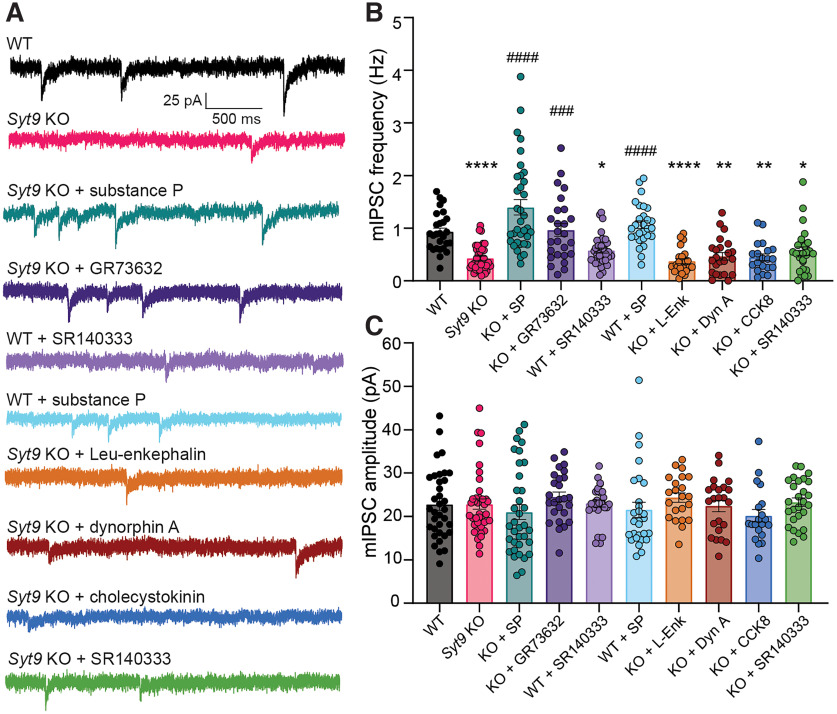
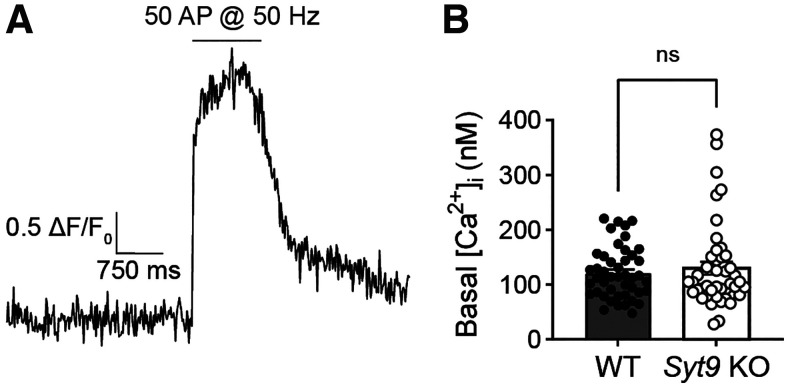
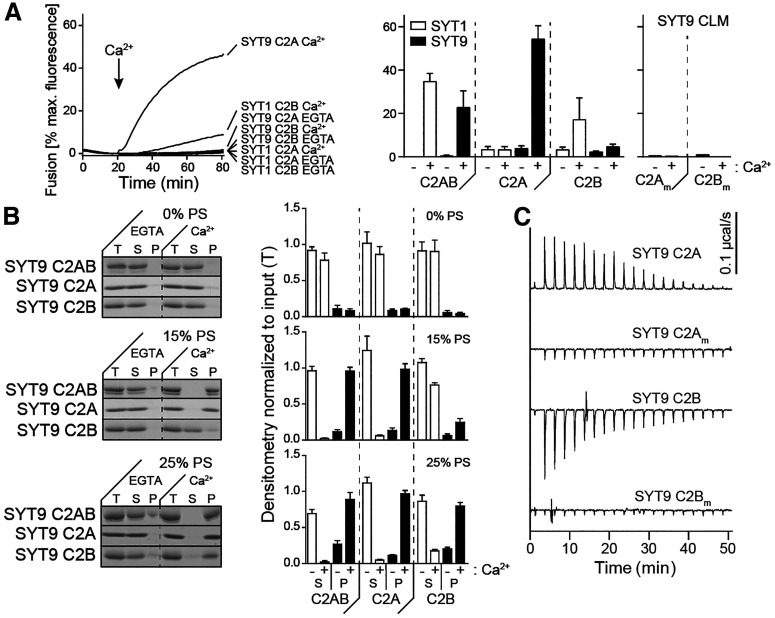
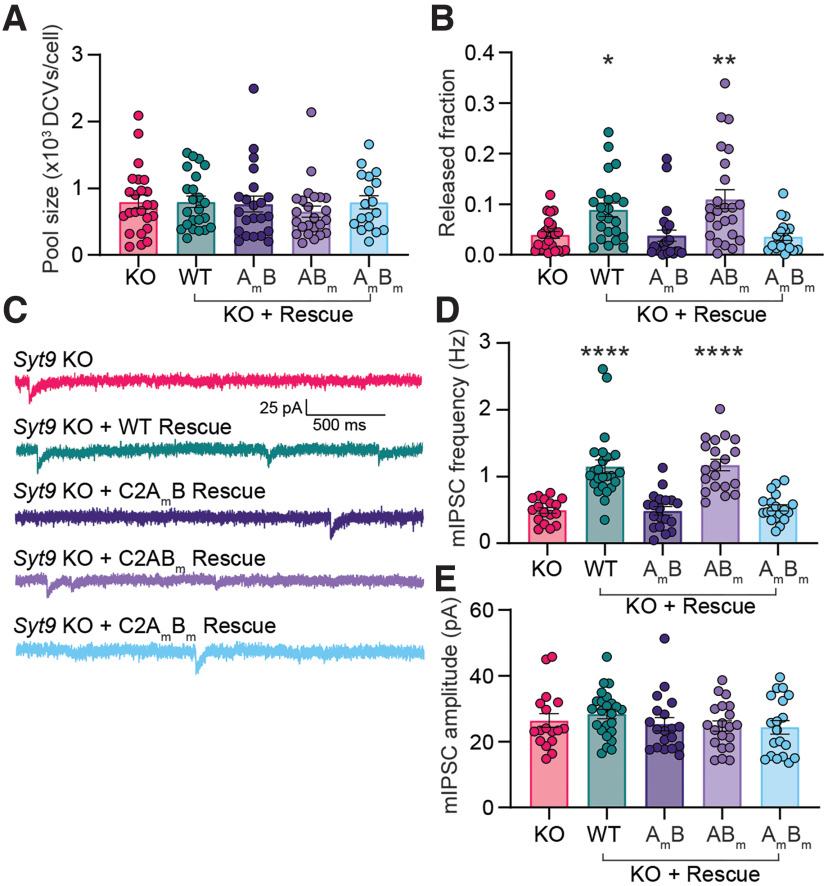
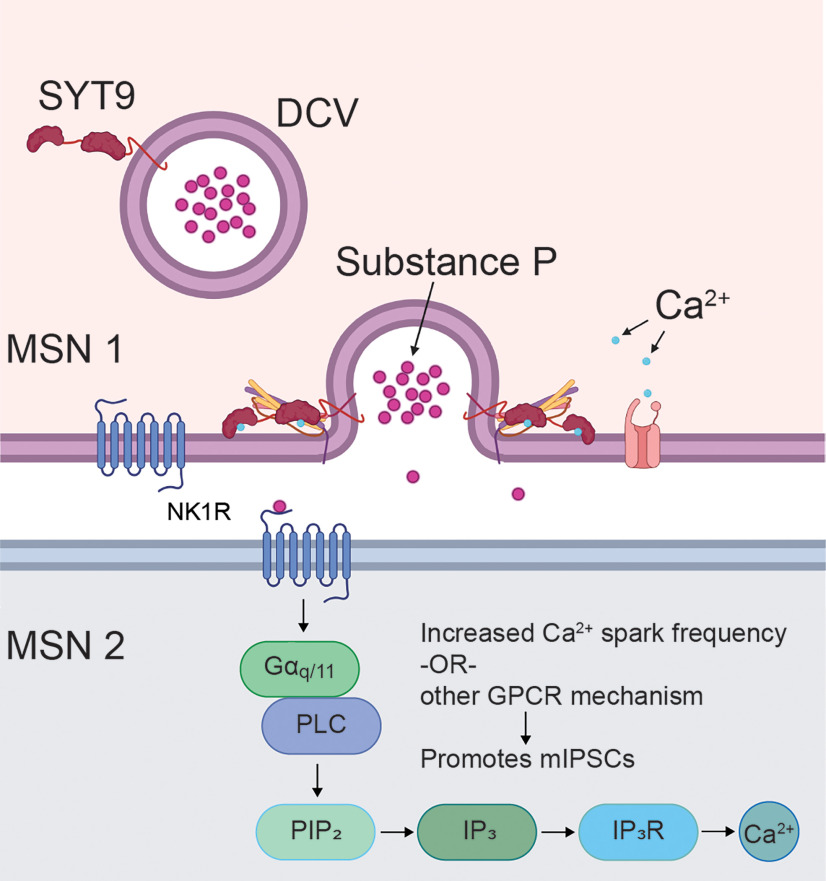
Synaptotagmin 9 (SYT9) is a tandem C2 domain Ca2+ sensor for exocytosis in neuroendocrine cells; its function in neurons remains unclear. Here, we show that, in mixed-sex cultures, SYT9 does not trigger rapid synaptic vesicle exocytosis in mouse cortical, hippocampal, or striatal neurons, unless it is massively overexpressed. In striatal neurons, loss of SYT9 reduced the frequency of spontaneous neurotransmitter release events (minis). We delved into the underlying mechanism and discovered that SYT9 was localized to dense-core vesicles that contain substance P (SP). Loss of SYT9 impaired SP release, causing the observed decrease in mini frequency. This model is further supported by loss of function mutants. Namely, Ca2+ binding to the C2A domain of SYT9 triggered membrane fusion in vitro, and mutations that disrupted this activity abolished the ability of SYT9 to regulate both SP release and mini frequency. We conclude that SYT9 indirectly regulates synaptic transmission in striatal neurons by controlling SP release.SIGNIFICANCE STATEMENT Synaptotagmin 9 (SYT9) has been described as a Ca2+ sensor for dense-core vesicle (DCV) exocytosis in neuroendocrine cells, but its role in neurons remains unclear, despite widespread expression in the brain. This article examines the role of SYT9 in synaptic transmission across cultured cortical, hippocampal, and striatal neuronal preparations. We found that SYT9 regulates spontaneous neurotransmitter release in striatal neurons by serving as a Ca2+ sensor for the release of the neuromodulator substance P from DCVs. This demonstrates a novel role for SYT9 in neurons and uncovers a new field of study into neuromodulation by SYT9, a protein that is widely expressed in the brain.
Keywords: exocytosis; neuromodulation; neuropeptide; spontaneous neurotransmitter release; synaptic transmission; synaptotagmin.
Copyright © 2023 the authors.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Allosteric stabilization of calcium and phosphoinositide dual binding engages several synaptotagmins in fast exocytosis.Elife. 2022 Aug 5;11:e74810. doi: 10.7554/eLife.74810. Elife. 2022. PMID: 35929728 Free PMC article.
-
Deconstructing Synaptotagmin-1's Distinct Roles in Synaptic Vesicle Priming and Neurotransmitter Release.J Neurosci. 2022 Apr 6;42(14):2856-2871. doi: 10.1523/JNEUROSCI.1945-21.2022. Epub 2022 Feb 22. J Neurosci. 2022. PMID: 35193927 Free PMC article.
-
Genetic analysis of synaptotagmin-7 function in synaptic vesicle exocytosis.Proc Natl Acad Sci U S A. 2008 Mar 11;105(10):3986-91. doi: 10.1073/pnas.0712372105. Epub 2008 Feb 28. Proc Natl Acad Sci U S A. 2008. PMID: 18308933 Free PMC article.
-
Fast, synchronous neurotransmitter release: Past, present and future.Neuroscience. 2020 Jul 15;439:22-27. doi: 10.1016/j.neuroscience.2019.04.030. Epub 2019 Apr 29. Neuroscience. 2020. PMID: 31047980 Review.
-
How does synaptotagmin trigger neurotransmitter release?Annu Rev Biochem. 2008;77:615-41. doi: 10.1146/annurev.biochem.77.062005.101135. Annu Rev Biochem. 2008. PMID: 18275379 Review.
Cited by
-
SYT1-Associated Neurodevelopmental Disorder: A Narrative Review.Children (Basel). 2022 Sep 22;9(10):1439. doi: 10.3390/children9101439. Children (Basel). 2022. PMID: 36291375 Free PMC article. Review.
-
Functional genomics screens reveal a role for TBC1D24 and SV2B in antibody-dependent enhancement of dengue virus infection.bioRxiv [Preprint]. 2024 Apr 27:2024.04.26.591029. doi: 10.1101/2024.04.26.591029. bioRxiv. 2024. Update in: J Virol. 2024 Nov 19;98(11):e0158224. doi: 10.1128/jvi.01582-24 PMID: 38712102 Free PMC article. Updated. Preprint.
-
Synaptotagmin-9 in mouse retina.Vis Neurosci. 2024 Sep 18;41:E003. doi: 10.1017/S0952523824000026. Vis Neurosci. 2024. PMID: 39291699 Free PMC article.
-
SYNAPTOTAGMIN-9 IN MOUSE RETINA.bioRxiv [Preprint]. 2024 Mar 16:2023.06.27.546758. doi: 10.1101/2023.06.27.546758. bioRxiv. 2024. Update in: Vis Neurosci. 2024 Sep 18;41:E003. doi: 10.1017/S0952523824000026 PMID: 37425946 Free PMC article. Updated. Preprint.
-
Exosomes in the Diagnosis of Neuropsychiatric Diseases: A Review.Biology (Basel). 2024 May 28;13(6):387. doi: 10.3390/biology13060387. Biology (Basel). 2024. PMID: 38927267 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous