

The unique structural characteristics of the Kir 7.1 inward rectifier potassium channel: a novel player in energy homeostasis control
- PMID: 36717105
- PMCID: PMC10026989
- DOI: 10.1152/ajpcell.00335.2022
The unique structural characteristics of the Kir 7.1 inward rectifier potassium channel: a novel player in energy homeostasis control
Abstract
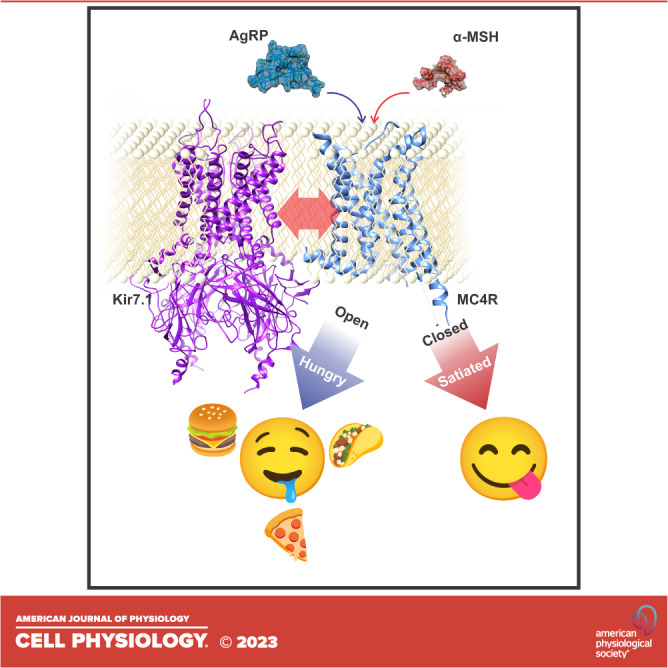
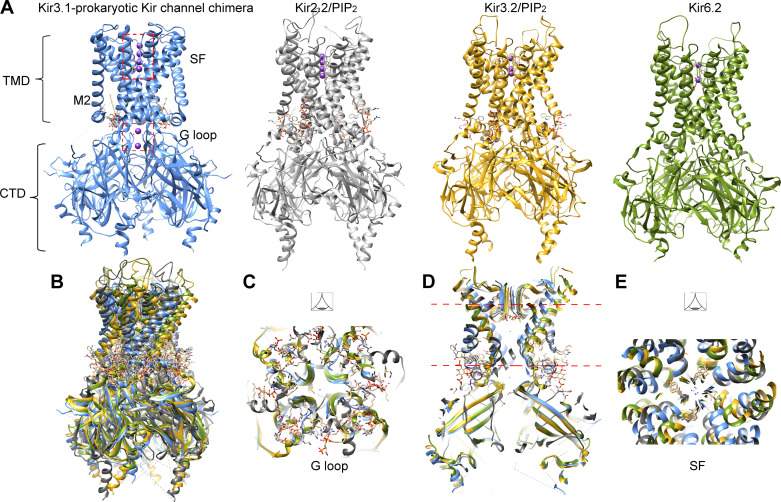
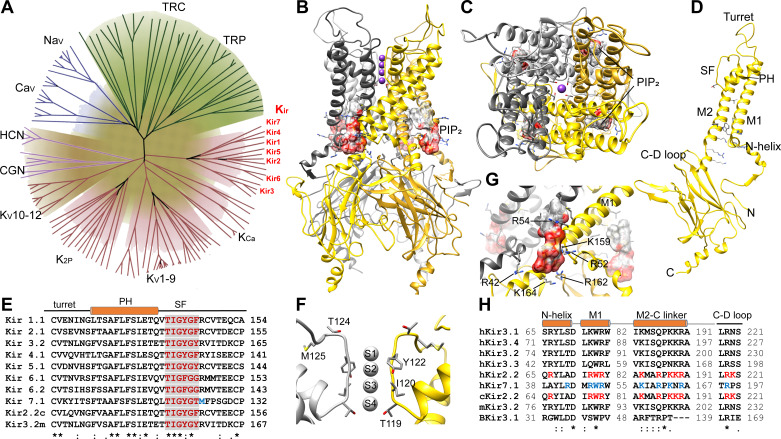
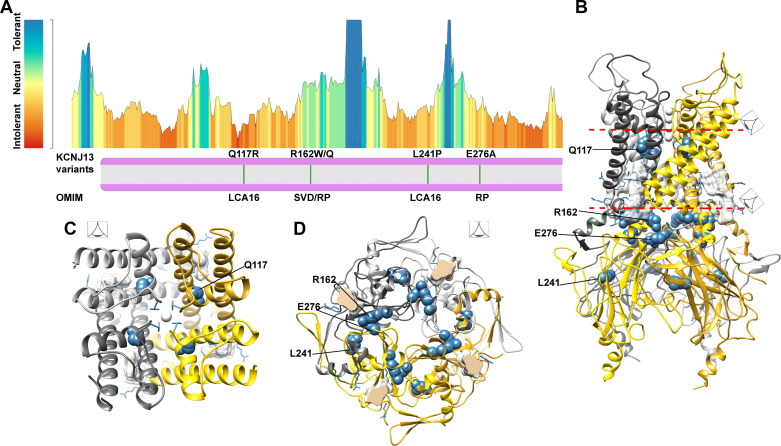
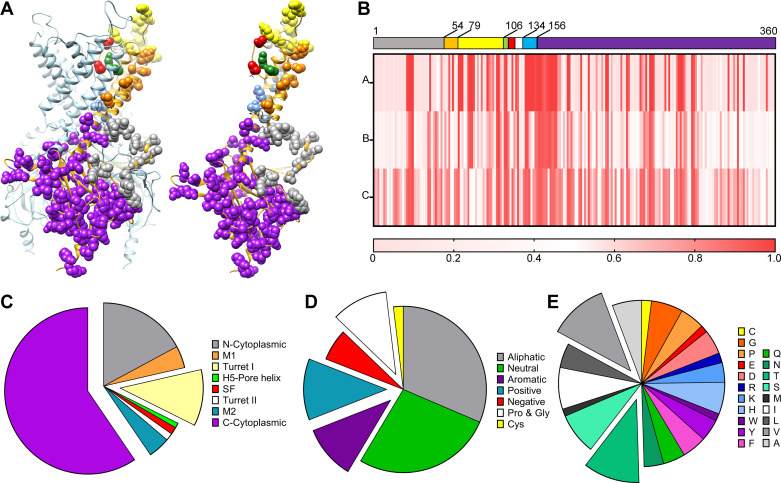
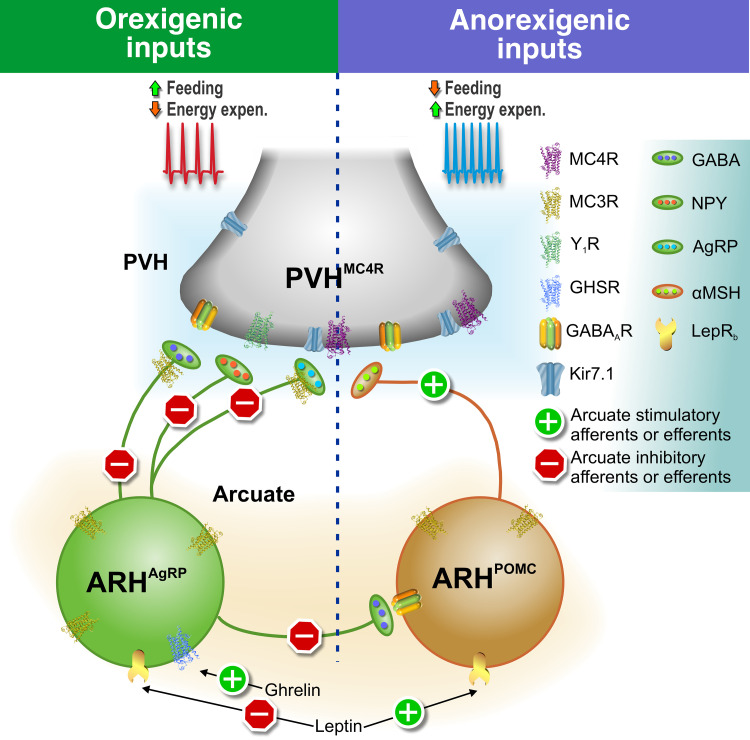
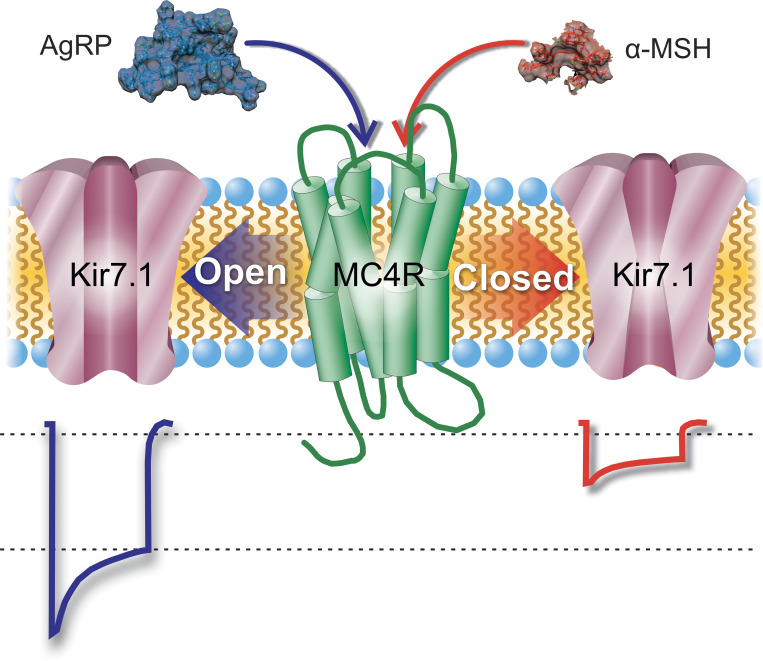
The inward rectifier potassium channel Kir7.1, encoded by the KCNJ13 gene, is a tetramer composed of two-transmembrane domain-spanning monomers, closer in homology to Kir channels associated with potassium transport such as Kir1.1, 1.2, and 1.3. Compared with other channels, Kir7.1 exhibits small unitary conductance and low dependence on external potassium. Kir7.1 channels also show a phosphatidylinositol 4,5-bisphosphate (PIP2) dependence for opening. Accordingly, retinopathy-associated Kir7.1 mutations mapped at the binding site for PIP2 resulted in channel gating defects leading to channelopathies such as snowflake vitreoretinal degeneration and Leber congenital amaurosis in blind patients. Lately, this channel's role in energy homeostasis was reported due to the direct interaction with the melanocortin type 4 receptor (MC4R) in the hypothalamus. As this channel seems to play a multipronged role in potassium homeostasis and neuronal excitability, we will discuss what is predicted from a structural viewpoint and its possible implications for hunger control.
Keywords: energy homeostasis; inward rectifier potassium channel 13 (Kir7.1); melanocortin receptor 4; phosphatidylinositol 4,5-bisphosphate; single-nucleotide polymorphism.
Conflict of interest statement
R. D. Cone and L. E. Gimenez have equity interests in Courage Therapeutics, Inc. and are inventors of intellectual property optioned to the company. R. D. Cone is a founder, board member, and SAB Chair of Courage Therapeutics, Inc. The other authors have no conflicts of interest, financial or otherwise, to disclose.
Figures
Similar articles
-
G protein-coupled receptors differentially regulate glycosylation and activity of the inwardly rectifying potassium channel Kir7.1.J Biol Chem. 2018 Nov 16;293(46):17739-17753. doi: 10.1074/jbc.RA118.003238. Epub 2018 Sep 26. J Biol Chem. 2018. PMID: 30257863 Free PMC article.
-
Focus on Kir7.1: physiology and channelopathy.Channels (Austin). 2014;8(6):488-95. doi: 10.4161/19336950.2014.959809. Channels (Austin). 2014. PMID: 25558901 Free PMC article. Review.
-
ML418: The First Selective, Sub-Micromolar Pore Blocker of Kir7.1 Potassium Channels.ACS Chem Neurosci. 2016 Jul 20;7(7):1013-23. doi: 10.1021/acschemneuro.6b00111. Epub 2016 May 24. ACS Chem Neurosci. 2016. PMID: 27184474 Free PMC article.
-
Conformational changes at cytoplasmic intersubunit interactions control Kir channel gating.J Biol Chem. 2017 Jun 16;292(24):10087-10096. doi: 10.1074/jbc.M117.785154. Epub 2017 Apr 26. J Biol Chem. 2017. PMID: 28446610 Free PMC article.
-
Inward rectifier potassium (Kir) channels in the retina: living our vision.Am J Physiol Cell Physiol. 2022 Sep 1;323(3):C772-C782. doi: 10.1152/ajpcell.00112.2022. Epub 2022 Aug 1. Am J Physiol Cell Physiol. 2022. PMID: 35912989 Free PMC article. Review.
Cited by
-
Special collection on inward rectifying K+ channels.Am J Physiol Cell Physiol. 2023 Mar 1;324(3):C603-C605. doi: 10.1152/ajpcell.00457.2022. Epub 2023 Jan 23. Am J Physiol Cell Physiol. 2023. PMID: 36689674 Free PMC article.
-
Updates on Rare Genetic Variants, Genetic Testing, and Gene Therapy in Individuals With Obesity.Curr Obes Rep. 2024 Sep;13(3):626-641. doi: 10.1007/s13679-024-00567-y. Epub 2024 Jun 1. Curr Obes Rep. 2024. PMID: 38822963 Review.
-
Kir7.1 knockdown and inhibition alter renal electrolyte handling but not the development of hypertension in Dahl salt-sensitive rats.Am J Physiol Renal Physiol. 2023 Aug 1;325(2):F177-F187. doi: 10.1152/ajprenal.00059.2023. Epub 2023 Jun 15. Am J Physiol Renal Physiol. 2023. PMID: 37318990 Free PMC article.
References
-
- Shaya D, Findeisen F, Abderemane-Ali F, Arrigoni C, Wong S, Nurva SR, Loussouarn G, Minor DL Jr.. Structure of a prokaryotic sodium channel pore reveals essential gating elements and an outer ion binding site common to eukaryotic channels. J Mol Biol 426: 467–483, 2014. doi:10.1016/j.jmb.2013.10.010. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials