

Time-of-Flight Fragmentation Spectra Generated by the Proteomic Analysis of Single Human Cells Do Not Exhibit Atypical Fragmentation Patterns
- PMID: 36700448
- PMCID: PMC10502792
- DOI: 10.1021/acs.jproteome.2c00715
Time-of-Flight Fragmentation Spectra Generated by the Proteomic Analysis of Single Human Cells Do Not Exhibit Atypical Fragmentation Patterns
Abstract
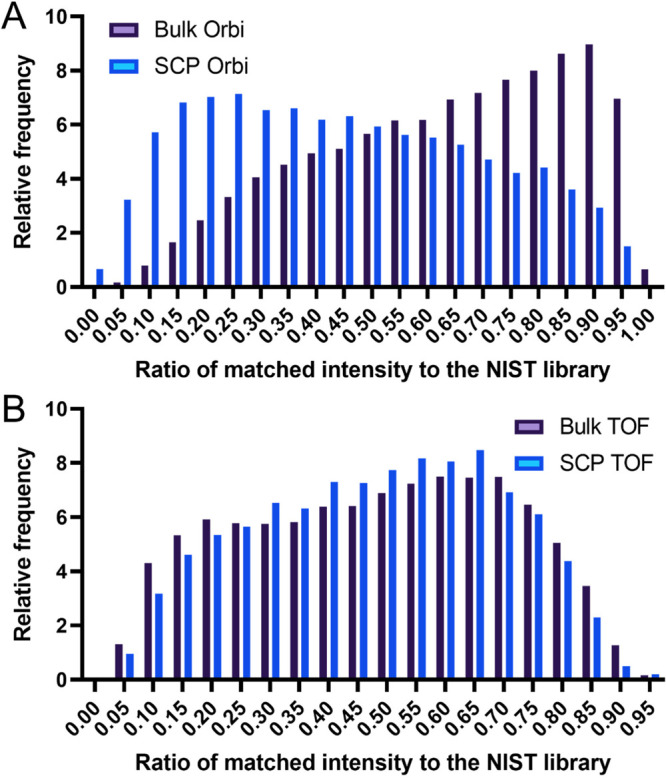
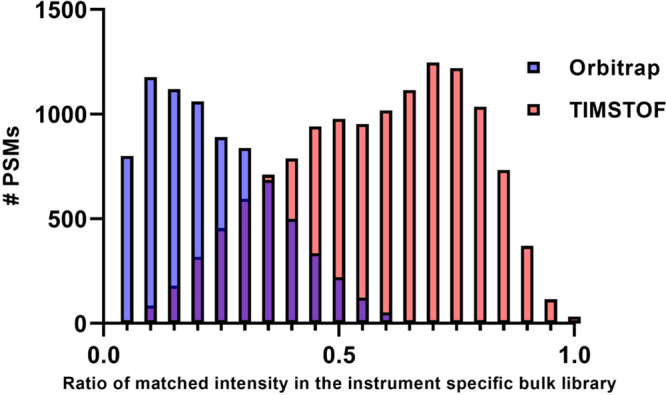
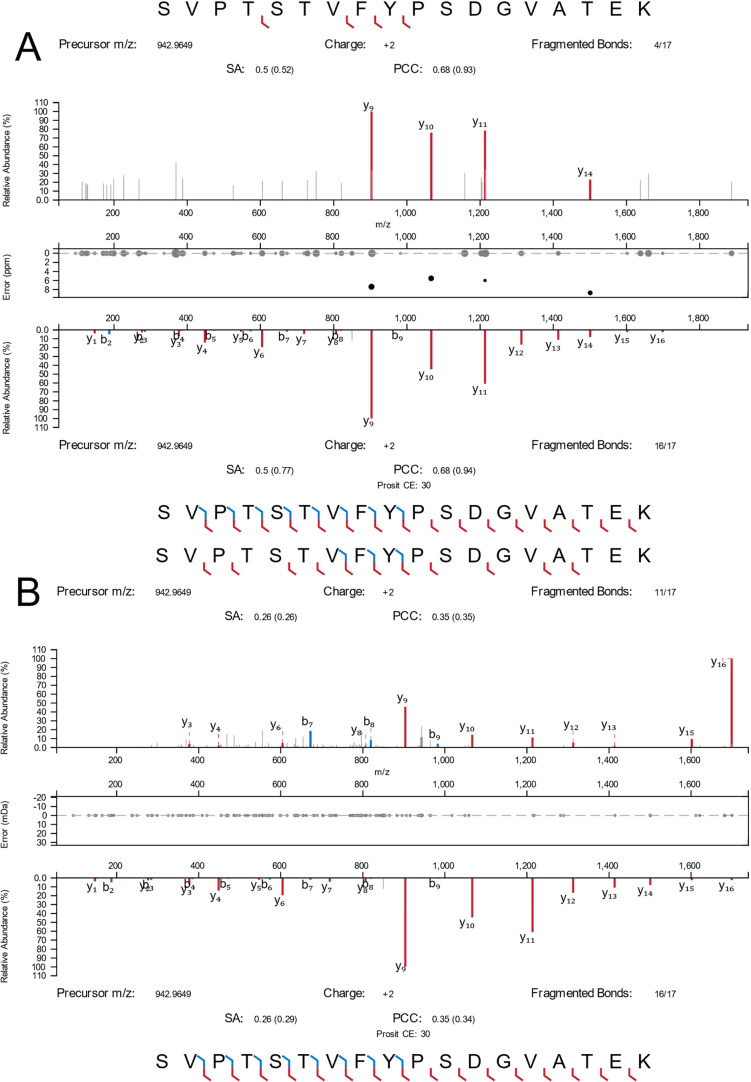
Recent work detailed the unique characteristics of fragmentation spectra derived from peptides from single human cells. This valuable report utilized an ultrahigh-field Orbitrap and directly compared the spectra obtained from high-concentration bulk cell HeLa lysates to those obtained from nanogram dilutions of the same and from nanowell-processed single HeLa cells. The analysis demonstrated marked differences between the fragmentation spectra generated at high and single-cell loads, most strikingly, the loss of high-mass y-series fragment ions. As significant differences exist in the physics of Orbitrap and time-of-flight mass analyzers, a comparison appeared warranted. A similar analysis was performed using isolated single pancreatic cancer cells compared to pools consisting of 100 cells. While a reanalysis of the prior Orbitrap data supports the author's original findings, the same trends are not observed in time-of-flight mass spectra of peptides from single human cells. The results are particularly striking when directly comparing the matched intensity fragment values between bulk and single-cell data generated on the same mass analyzers. Instrument acquisition files, processed data, and spectrum libraries are publicly available on MASSIVE via accession MSV000090635.
Keywords: Orbitrap single cell; TIMSTOF single cell; single-cell proteomics.
Conflict of interest statement
The author declares no competing financial interest.
Figures
Similar articles
-
Statistical characterization of HCD fragmentation patterns of tryptic peptides on an LTQ Orbitrap Velos mass spectrometer.J Proteomics. 2014 Sep 23;109:26-37. doi: 10.1016/j.jprot.2014.06.012. Epub 2014 Jun 27. J Proteomics. 2014. PMID: 24981973
-
Collision energies on QTof and Orbitrap instruments: How to make proteomics measurements comparable?J Mass Spectrom. 2021 Jan;56(1):e4693. doi: 10.1002/jms.4693. Epub 2020 Dec 5. J Mass Spectrom. 2021. PMID: 33277714
-
Improving SRM assay development: a global comparison between triple quadrupole, ion trap, and higher energy CID peptide fragmentation spectra.J Proteome Res. 2011 Sep 2;10(9):4334-41. doi: 10.1021/pr200156b. Epub 2011 Jul 21. J Proteome Res. 2011. PMID: 21726076
-
High-energy collision induced dissociation of biomolecules: MALDI-TOF/RTOF mass spectrometry in comparison to tandem sector mass spectrometry.Comb Chem High Throughput Screen. 2009 Feb;12(2):137-55. doi: 10.2174/138620709787315436. Comb Chem High Throughput Screen. 2009. PMID: 19199883 Review.
-
Tandem mass spectral libraries of peptides and their roles in proteomics research.Mass Spectrom Rev. 2017 Sep;36(5):634-648. doi: 10.1002/mas.21512. Epub 2016 Jul 12. Mass Spectrom Rev. 2017. PMID: 27403644 Review.
Cited by
-
Fragment ion intensity prediction improves the identification rate of non-tryptic peptides in timsTOF.Nat Commun. 2024 May 10;15(1):3956. doi: 10.1038/s41467-024-48322-0. Nat Commun. 2024. PMID: 38730277 Free PMC article.
-
Challenges and Opportunities for Single-cell Computational Proteomics.Mol Cell Proteomics. 2023 Apr;22(4):100518. doi: 10.1016/j.mcpro.2023.100518. Epub 2023 Feb 23. Mol Cell Proteomics. 2023. PMID: 36828128 Free PMC article. Review.
-
Analyzing Posttranslational Modifications in Single Cells.Methods Mol Biol. 2024;2817:145-156. doi: 10.1007/978-1-0716-3934-4_12. Methods Mol Biol. 2024. PMID: 38907153 Review.
-
MSBooster: improving peptide identification rates using deep learning-based features.Nat Commun. 2023 Jul 27;14(1):4539. doi: 10.1038/s41467-023-40129-9. Nat Commun. 2023. PMID: 37500632 Free PMC article.
References
-
- Kelly R.; Zhu Y.; Liang Y.; Cong Y.; Piehowski P.; Dou M.; Zhao R.; Qian W.-J.; Burnum-Johnson K.; Ansong C. Single Cell Proteome Mapping of Tissue Heterogeneity Using Microfluidic Nanodroplet Sample Processing and Ultrasensitive LC-MS. J. Biomol. Technol. 2019, 30, S61.
-
- Hartlmayr D.; Ctortecka C.; Seth A.; Mendjan S.; Tourniaire G.; Mechtler K.. An Automated Workflow for Label-Free and Multiplexed Single Cell Proteomics Sample Preparation at Unprecedented Sensitivity. bioRxiv 2021, 10.1101/2021.04.14.439828. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
