

Rilotumumab Resistance Acquired by Intracrine Hepatocyte Growth Factor Signaling
- PMID: 36672409
- PMCID: PMC9857108
- DOI: 10.3390/cancers15020460
Rilotumumab Resistance Acquired by Intracrine Hepatocyte Growth Factor Signaling
Abstract
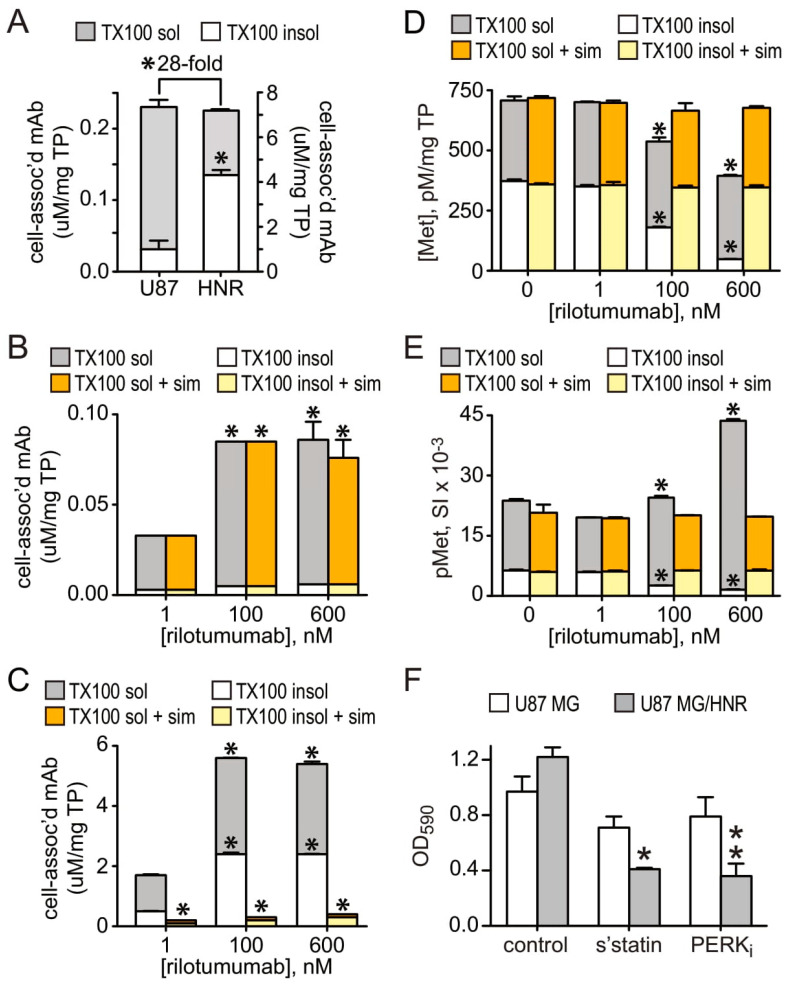
Drug resistance is a long-standing impediment to effective systemic cancer therapy and acquired drug resistance is a growing problem for molecularly-targeted therapeutics that otherwise have shown unprecedented successes in disease control. The hepatocyte growth factor (HGF)/Met receptor pathway signaling is frequently involved in cancer and has been a subject of targeted drug development for nearly 30 years. To anticipate and study specific resistance mechanisms associated with targeting this pathway, we engineered resistance to the HGF-neutralizing antibody rilotumumab in glioblastoma cells harboring autocrine HGF/Met signaling, a frequent abnormality of this brain cancer in humans. We found that rilotumumab resistance was acquired through an unusual mechanism comprising dramatic HGF overproduction and misfolding, endoplasmic reticulum (ER) stress-response signaling and redirected vesicular trafficking that effectively sequestered rilotumumab and misfolded HGF from native HGF and activated Met. Amplification of MET and HGF genes, with evidence of rapidly acquired intron-less, reverse-transcribed copies in DNA, was also observed. These changes enabled persistent Met pathway activation and improved cell survival under stress conditions. Point mutations in the HGF pathway or other complementary or downstream growth regulatory cascades that are frequently associated with targeted drug resistance in other prevalent cancer types were not observed. Although resistant cells were significantly more malignant, they retained sensitivity to Met kinase inhibition and acquired sensitivity to inhibition of ER stress signaling and cholesterol biosynthesis. Defining this mechanism reveals details of a rapidly acquired yet highly-orchestrated multisystem route of resistance to a selective molecularly-targeted agent and suggests strategies for early detection and effective intervention.
Keywords: Met; acquired drug resistance; glioblastoma; hepatocyte growth factor; rilotumumab.
Conflict of interest statement
Karen Rex, Joanna Schmidt, Daniel Baker, Michael A. Damore, Angela Coxon, and Teresa L. Burgess are current or former employees of Amgen Inc. Authors declare that there are no conflicts of interest.
Figures
Similar articles
-
Hepatocyte growth factor/MET in cancer progression and biomarker discovery.Cancer Sci. 2017 Mar;108(3):296-307. doi: 10.1111/cas.13156. Cancer Sci. 2017. PMID: 28064454 Free PMC article. Review.
-
Rilotumumab, a mAb against human hepatocyte growth factor for the treatment of cancer.Curr Opin Mol Ther. 2009 Aug;11(4):448-55. Curr Opin Mol Ther. 2009. PMID: 19649990
-
Emerging molecular targets in oncology: clinical potential of MET/hepatocyte growth-factor inhibitors.Onco Targets Ther. 2014 Jun 12;7:1001-14. doi: 10.2147/OTT.S44941. eCollection 2014. Onco Targets Ther. 2014. PMID: 24959087 Free PMC article. Review.
-
Role of HGF/MET axis in resistance of lung cancer to contemporary management.Transl Lung Cancer Res. 2012 Sep;1(3):179-93. doi: 10.3978/j.issn.2218-6751.2012.09.04. Transl Lung Cancer Res. 2012. PMID: 25806180 Free PMC article. Review.
-
Differential responses of MET activations to MET kinase inhibitor and neutralizing antibody.J Transl Med. 2018 Sep 12;16(1):253. doi: 10.1186/s12967-018-1628-y. J Transl Med. 2018. PMID: 30208970 Free PMC article.
Cited by
-
PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer.Mol Cancer. 2023 Aug 18;22(1):138. doi: 10.1186/s12943-023-01827-6. Mol Cancer. 2023. PMID: 37596643 Free PMC article. Review.
-
The Different Roles of MET in the Development and Treatment of Cancer.Cancers (Basel). 2023 Oct 21;15(20):5087. doi: 10.3390/cancers15205087. Cancers (Basel). 2023. PMID: 37894454 Free PMC article.
-
A real-world pharmacovigilance study of FDA adverse event reporting system events for Capmatinib.Sci Rep. 2024 May 18;14(1):11388. doi: 10.1038/s41598-024-62356-w. Sci Rep. 2024. PMID: 38762672 Free PMC article.
-
Receptor tyrosine kinases: biological functions and anticancer targeted therapy.MedComm (2020). 2023 Dec 7;4(6):e446. doi: 10.1002/mco2.446. eCollection 2023 Dec. MedComm (2020). 2023. PMID: 38077251 Free PMC article. Review.
References
-
- Bean J., Brennan C., Shih J.Y., Riely G., Viale A., Wang L., Chitale D., Motoi N., Szoke J., Broderick S., et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. - DOI - PMC - PubMed
-
- Suda K., Murakami I., Katayama T., Tomizawa K., Osada H., Sekido Y., Maehara Y., Yatabe Y., Mitsudomi T. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin. Cancer Res. 2010;16:5489–5498. doi: 10.1158/1078-0432.CCR-10-1371. - DOI - PubMed
-
- Yano S., Wang W., Li Q., Matsumoto K., Sakurama H., Nakamura T., Ogino H., Kakiuchi S., Hanibuchi M., Nishioka Y., et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. - DOI - PubMed
-
- Yano S., Yamada T., Takeuchi S., Tachibana K., Minami Y., Yatabe Y., Mitsudomi T., Tanaka H., Kimura T., Kudoh S., et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J. Thorac. Oncol. 2011;6:2011–2017. doi: 10.1097/JTO.0b013e31823ab0dd. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
