

Airway Smooth Muscle Regulated by Oxidative Stress in COPD
- PMID: 36671004
- PMCID: PMC9854973
- DOI: 10.3390/antiox12010142
Airway Smooth Muscle Regulated by Oxidative Stress in COPD
Abstract
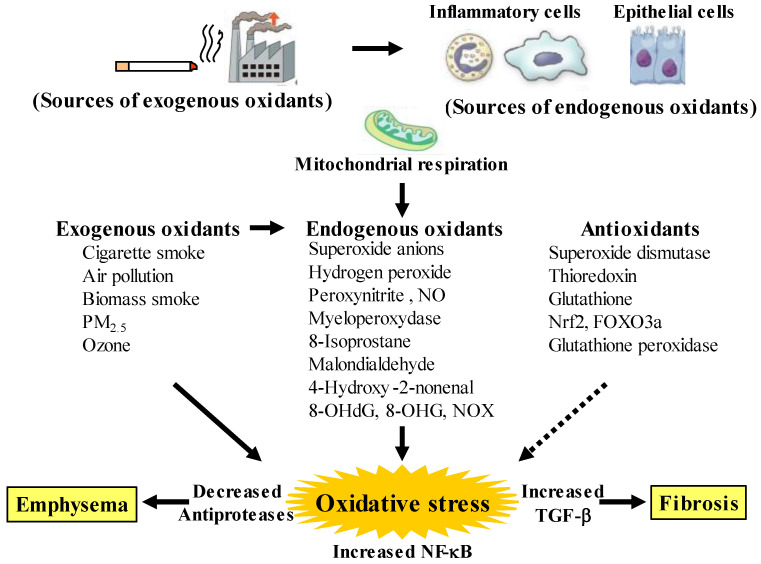
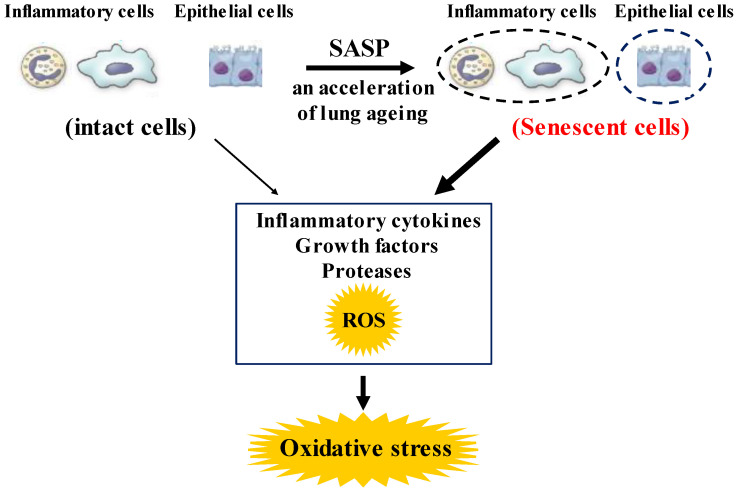
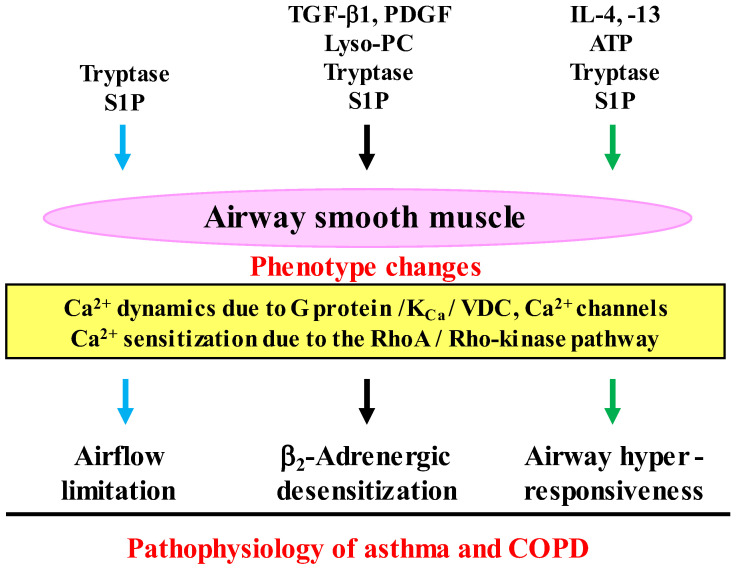
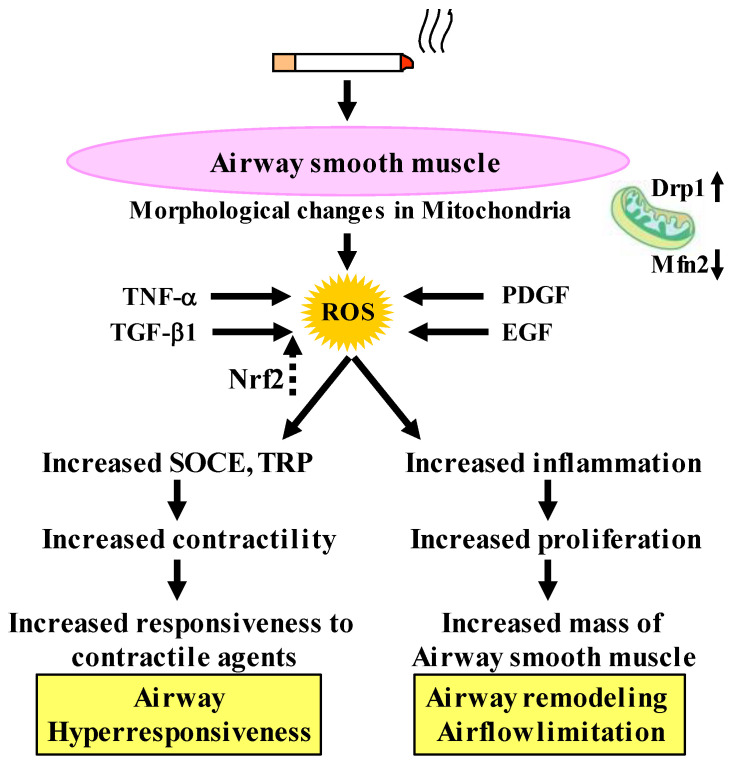
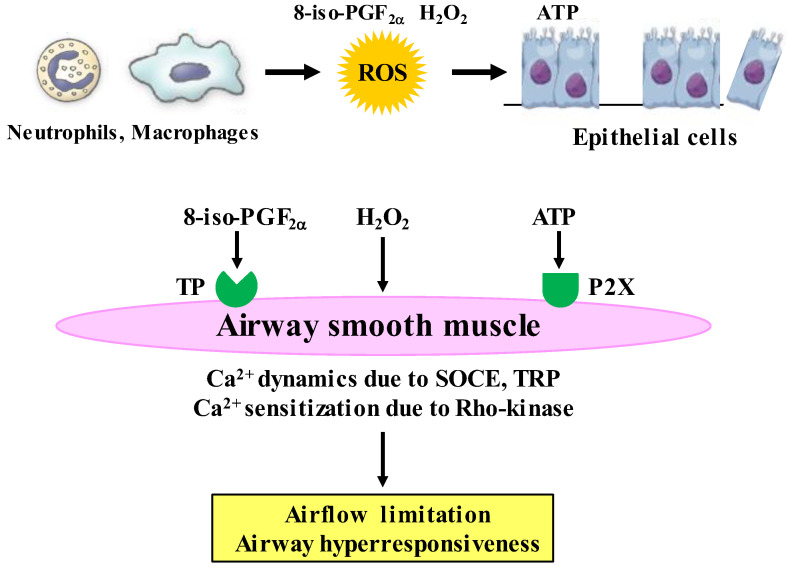
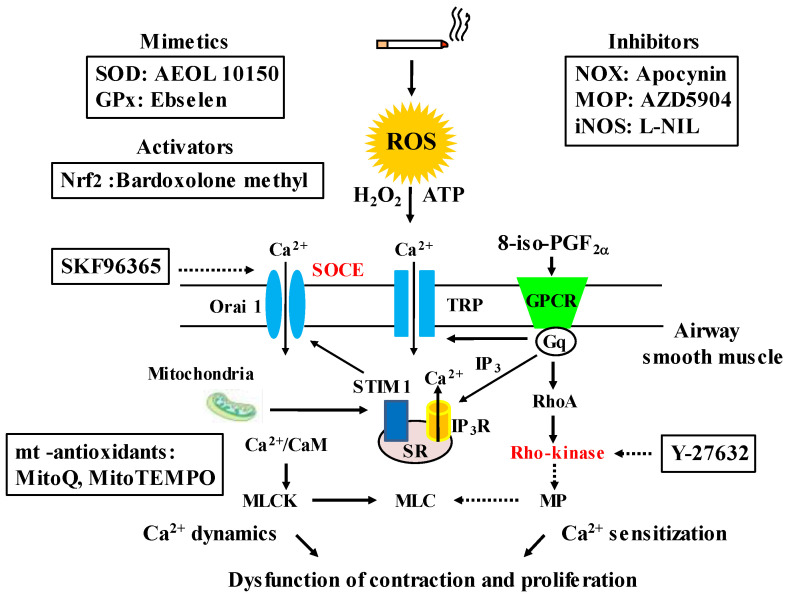
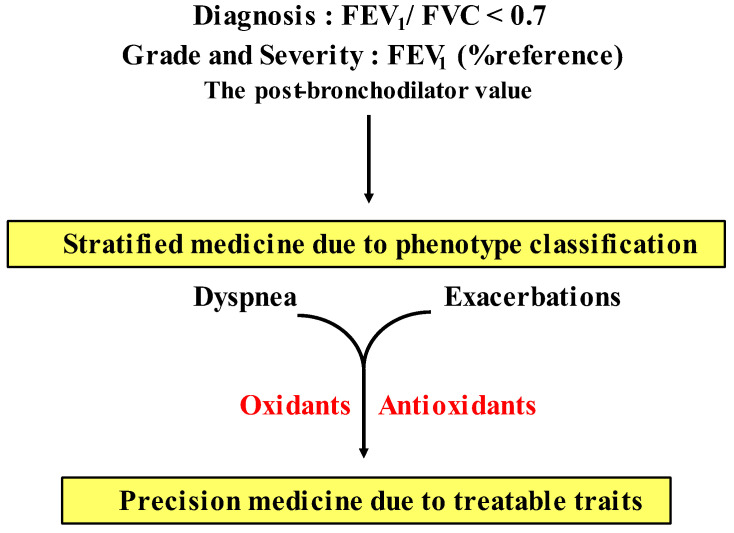
Since COPD is a heterogeneous disease, a specific anti-inflammatory therapy for this disease has not been established yet. Oxidative stress is recognized as a major predisposing factor to COPD related inflammatory responses, resulting in pathological features of small airway fibrosis and emphysema. However, little is known about effects of oxidative stress on airway smooth muscle. Cigarette smoke increases intracellular Ca2+ concentration and enhances response to muscarinic agonists in human airway smooth muscle. Cigarette smoke also enhances proliferation of these cells with altered mitochondrial protein. Hydrogen peroxide and 8-isoprostans are increased in the exhaled breath condensate in COPD. These endogenous oxidants cause contraction of tracheal smooth muscle with Ca2+ dynamics through Ca2+ channels and with Ca2+ sensitization through Rho-kinase. TNF-α and growth factors potentiate proliferation of these cells by synthesis of ROS. Oxidative stress can alter the function of airway smooth muscle through Ca2+ signaling. These phenotype changes are associated with manifestations (dyspnea, wheezing) and pathophysiology (airflow limitation, airway remodeling, airway hyperresponsiveness). Therefore, airway smooth muscle is a therapeutic target against COPD; oxidative stress should be included in treatable traits for COPD to advance precision medicine. Research into Ca2+ signaling related to ROS may contribute to the development of a novel agent for COPD.
Keywords: Ca2+ dynamics; Ca2+ sensitization; antioxidants; oxidants; phenotype changes; reactive oxygen species; tracheal smooth muscle.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Role of Airway Smooth Muscle in Inflammation Related to Asthma and COPD.Adv Exp Med Biol. 2021;1303:139-172. doi: 10.1007/978-3-030-63046-1_9. Adv Exp Med Biol. 2021. PMID: 33788192
-
Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease.J Allergy Clin Immunol. 2015 Sep;136(3):769-80. doi: 10.1016/j.jaci.2015.01.046. Epub 2015 Mar 29. J Allergy Clin Immunol. 2015. PMID: 25828268 Free PMC article.
-
Direct effects of hydrogen peroxide on airway smooth muscle tone: roles of Ca2+ influx and Rho-kinase.Eur J Pharmacol. 2007 Feb 5;556(1-3):151-6. doi: 10.1016/j.ejphar.2006.11.007. Epub 2006 Nov 10. Eur J Pharmacol. 2007. PMID: 17157292
-
Stressed out - The role of oxidative stress in airway smooth muscle dysfunction in asthma and COPD.Free Radic Biol Med. 2022 May 20;185:97-119. doi: 10.1016/j.freeradbiomed.2022.04.011. Epub 2022 Apr 23. Free Radic Biol Med. 2022. PMID: 35472411 Review.
-
The role of RhoA-mediated Ca2+ sensitization of bronchial smooth muscle contraction in airway hyperresponsiveness.J Smooth Muscle Res. 2004 Oct;40(4-5):155-67. doi: 10.1540/jsmr.40.155. J Smooth Muscle Res. 2004. PMID: 15655303 Review.
Cited by
-
Immunometabolism changes in fibrosis: from mechanisms to therapeutic strategies.Front Pharmacol. 2023 Jul 27;14:1243675. doi: 10.3389/fphar.2023.1243675. eCollection 2023. Front Pharmacol. 2023. PMID: 37576819 Free PMC article. Review.
-
Epicardial adipose tissue in patients with chronic obstructive pulmonary disease: systematic review with meta‑analysis and trial sequential analysis.BMC Pulm Med. 2023 Jul 3;23(1):241. doi: 10.1186/s12890-023-02535-z. BMC Pulm Med. 2023. PMID: 37400821 Free PMC article.
-
Involvement of Lysophospholipids in Pulmonary Vascular Functions and Diseases.Biomedicines. 2024 Jan 8;12(1):124. doi: 10.3390/biomedicines12010124. Biomedicines. 2024. PMID: 38255229 Free PMC article. Review.
-
Decoding LncRNA in COPD: Unveiling Prognostic and Diagnostic Power and Their Driving Role in Lung Cancer Progression.Int J Mol Sci. 2024 Aug 19;25(16):9001. doi: 10.3390/ijms25169001. Int J Mol Sci. 2024. PMID: 39201688 Free PMC article. Review.
-
Metabolic pathways in immune senescence and inflammaging: Novel therapeutic strategy for chronic inflammatory lung diseases. An EAACI position paper from the Task Force for Immunopharmacology.Allergy. 2024 May;79(5):1089-1122. doi: 10.1111/all.15977. Epub 2023 Dec 18. Allergy. 2024. PMID: 38108546 Free PMC article. Review.
References
-
- Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for Prevention, Diagnosis and Management of Chronic Obstructive Pulmonary Disease: 2023 Report. [(accessed on 1 December 2022)]. Available online: https://goldcopd.org/
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
