

The Role of Oxidants in Percutaneous Coronary Intervention-Induced Endothelial Dysfunction: Can We Harness Redox Signaling to Improve Clinical Outcomes?
- PMID: 36641638
- PMCID: PMC10402708
- DOI: 10.1089/ars.2022.0204
The Role of Oxidants in Percutaneous Coronary Intervention-Induced Endothelial Dysfunction: Can We Harness Redox Signaling to Improve Clinical Outcomes?
Abstract
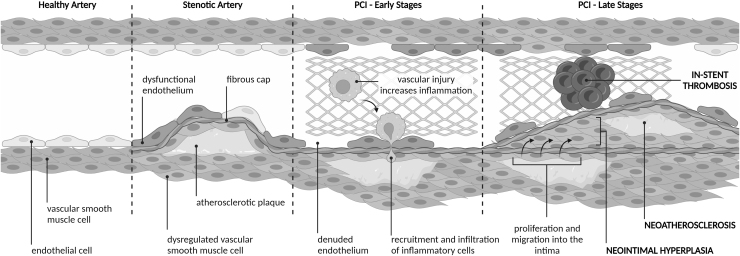
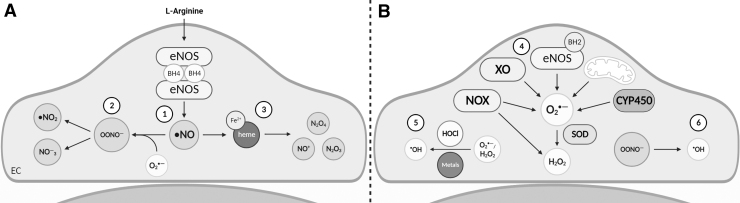
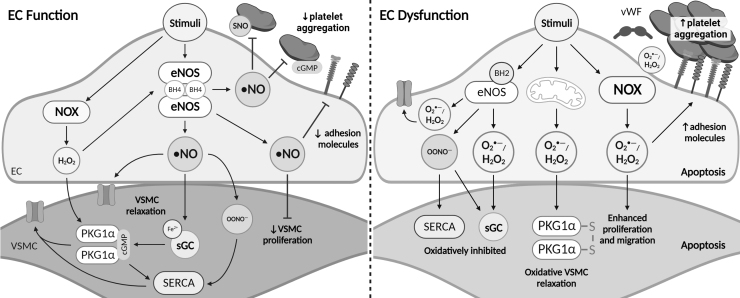
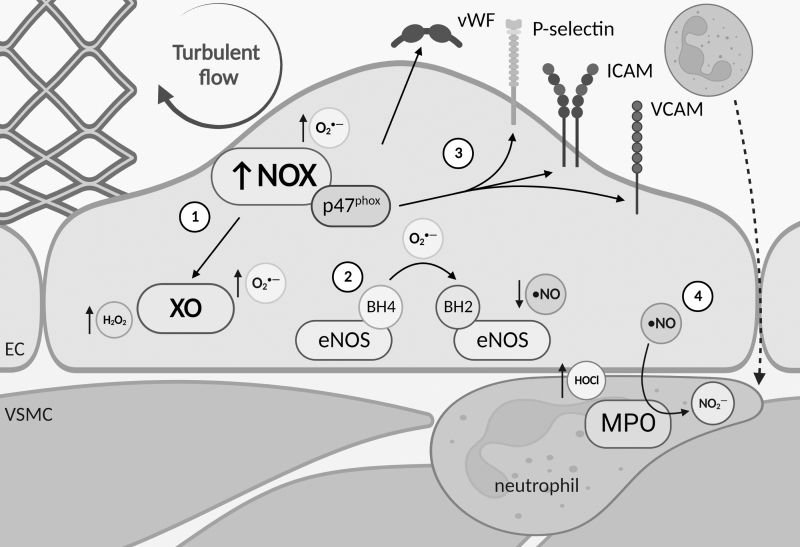
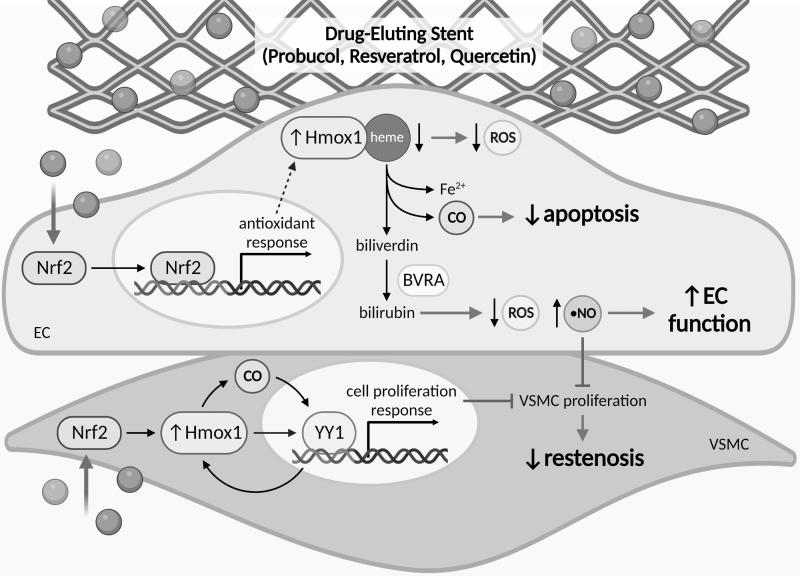
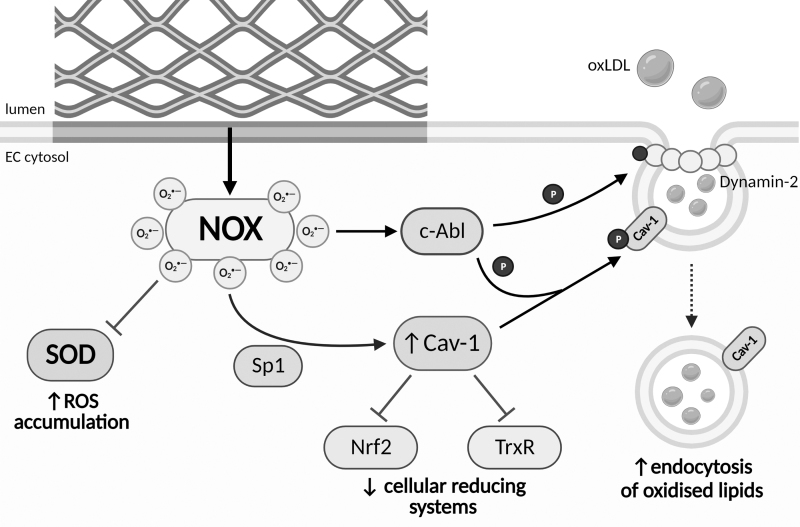
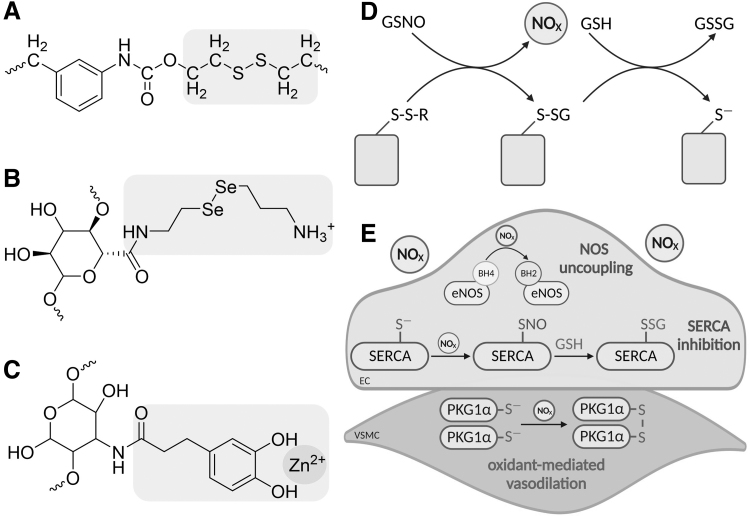
Significance: Coronary artery disease (CAD) is commonly treated using percutaneous coronary interventions (PCI). However, PCI with stent placement damages the endothelium, and failure to restore endothelial function may result in PCI failure with poor patient outcomes. Recent Advances: Oxidative signaling is central to maintaining endothelial function. Potentiation of oxidant production, as observed post-PCI, results in endothelial dysfunction (ED). This review delves into our current understanding of the physiological role that endothelial-derived oxidants play within the vasculature and the effects of altered redox signaling during dysfunction. We then examine the impact of PCI and intracoronary stent placement on oxidant production in the endothelium, which can culminate in stent failure. Finally, we explore how recent advances in PCI and stent technologies aim to mitigate PCI-induced oxidative damage and improve clinical outcomes. Critical Issues: Current PCI technologies exacerbate cellular oxidant levels, driving ED. If left uncontrolled, oxidative signaling leads to increased intravascular inflammation, restenosis, and neoatherosclerosis. Future Directions: Through the development of novel biomaterials and therapeutics, we can limit PCI-induced oxidant production, allowing for the restoration of a healthy endothelium and preventing CAD recurrence.
Keywords: CAD; PCI; endothelial dysfunction; oxidants; redox signaling.
Conflict of interest statement
No competing financial interests exist.
Figures
Similar articles
-
Risk factors of in-stent restenosis among coronary artery disease patients with syphilis undergoing percutaneous coronary intervention: a retrospective study.BMC Cardiovasc Disord. 2021 Sep 15;21(1):438. doi: 10.1186/s12872-021-02245-6. BMC Cardiovasc Disord. 2021. PMID: 34525967 Free PMC article.
-
Angiotensin type 1 receptor A1166C gene polymorphism is associated with endothelial dysfunction and in-stent restenosis after percutaneous coronary intervention.Int J Clin Exp Pathol. 2015 Jun 1;8(6):7350-7. eCollection 2015. Int J Clin Exp Pathol. 2015. PMID: 26261635 Free PMC article.
-
Percutaneous coronary intervention among patients with left ventricular systolic dysfunction: a review and meta-analysis of 19 clinical studies.Coron Artery Dis. 2012 Nov;23(7):469-79. doi: 10.1097/MCA.0b013e3283587804. Coron Artery Dis. 2012. PMID: 22960383 Review.
-
Role of endothelial dysfunction in determining angina after percutaneous coronary intervention: Learning from pathophysiology to optimize treatment.Prog Cardiovasc Dis. 2020 May-Jun;63(3):233-242. doi: 10.1016/j.pcad.2020.02.009. Epub 2020 Feb 13. Prog Cardiovasc Dis. 2020. PMID: 32061633 Review.
-
Coronary Artery Bypass Surgery Versus Drug-Eluting Stent Implantation for Left Main or Multivessel Coronary Artery Disease: A Meta-Analysis of Individual Patient Data.JACC Cardiovasc Interv. 2016 Dec 26;9(24):2481-2489. doi: 10.1016/j.jcin.2016.10.008. JACC Cardiovasc Interv. 2016. PMID: 28007199
Cited by
-
The role of hemoglobin A1c as a predictor of major adverse cardiovascular events in patients with type 2 diabetes mellitus after percutaneous coronary intervention: a case-cohort study.BMC Cardiovasc Disord. 2024 Oct 22;24(1):583. doi: 10.1186/s12872-024-04267-2. BMC Cardiovasc Disord. 2024. PMID: 39438790 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
