

Regression of cardiac hypertrophy in health and disease: mechanisms and therapeutic potential
- PMID: 36596855
- PMCID: PMC10121965
- DOI: 10.1038/s41569-022-00806-6
Regression of cardiac hypertrophy in health and disease: mechanisms and therapeutic potential
Abstract
Left ventricular hypertrophy is a leading risk factor for cardiovascular morbidity and mortality. Although reverse ventricular remodelling was long thought to be irreversible, evidence from the past three decades indicates that this process is possible with many existing heart disease therapies. The regression of pathological hypertrophy is associated with improved cardiac function, quality of life and long-term health outcomes. However, less than 50% of patients respond favourably to most therapies, and the reversibility of remodelling is influenced by many factors, including age, sex, BMI and disease aetiology. Cardiac hypertrophy also occurs in physiological settings, including pregnancy and exercise, although in these cases, hypertrophy is associated with normal or improved ventricular function and is completely reversible postpartum or with cessation of training. Studies over the past decade have identified the molecular features of hypertrophy regression in health and disease settings, which include modulation of protein synthesis, microRNAs, metabolism and protein degradation pathways. In this Review, we summarize the evidence for hypertrophy regression in patients with current first-line pharmacological and surgical interventions. We further discuss the molecular features of reverse remodelling identified in cell and animal models, highlighting remaining knowledge gaps and the essential questions for future investigation towards the goal of designing specific therapies to promote regression of pathological hypertrophy.
© 2023. Springer Nature Limited.
Conflict of interest statement
Competing interests
L.A.L. is a co-founder of MyoKardia, acquired by Bristol Myers Squib, and is a paid member of their Scientific Advisory Board. The other authors declare no competing interests.
Figures
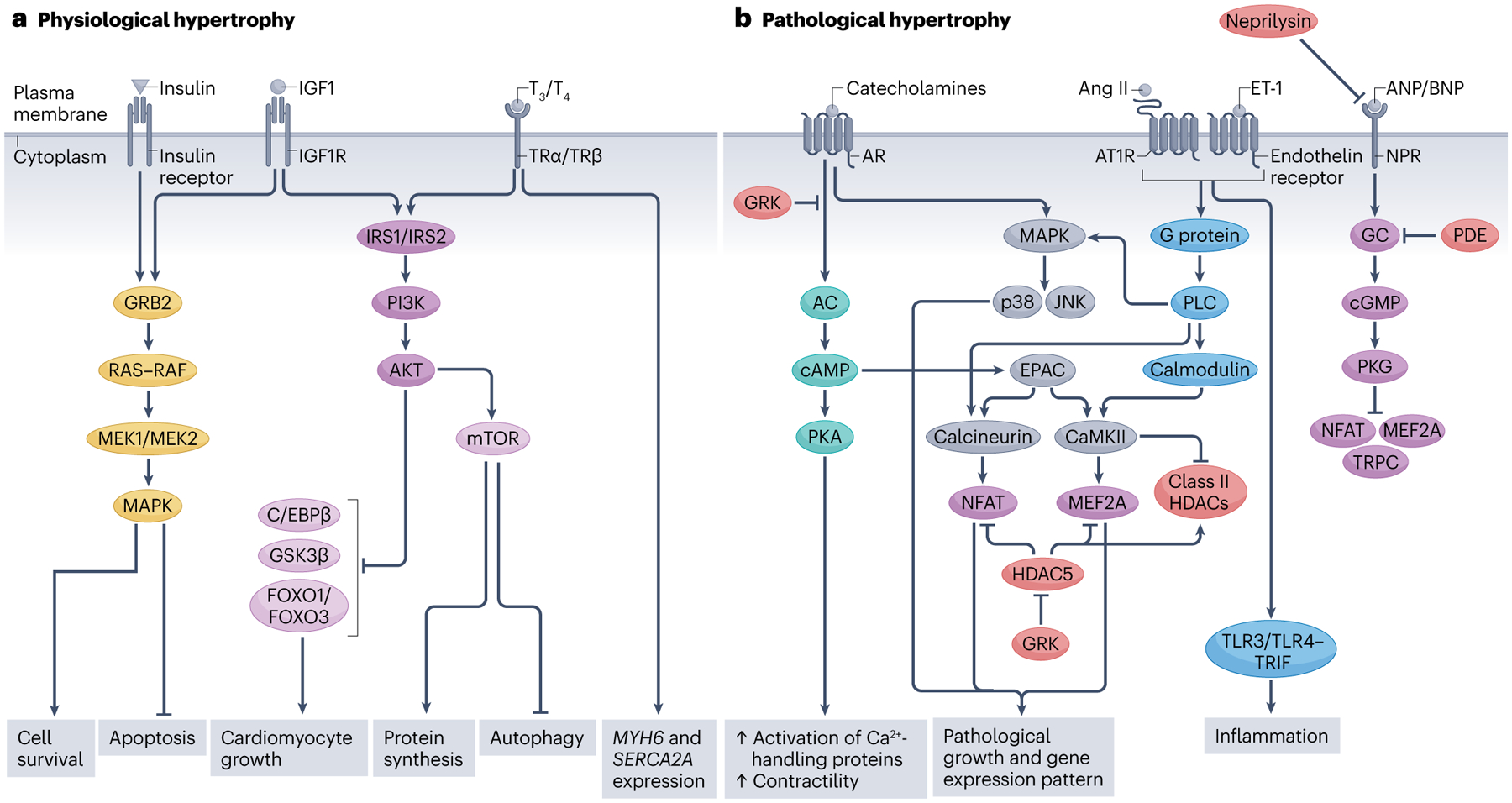
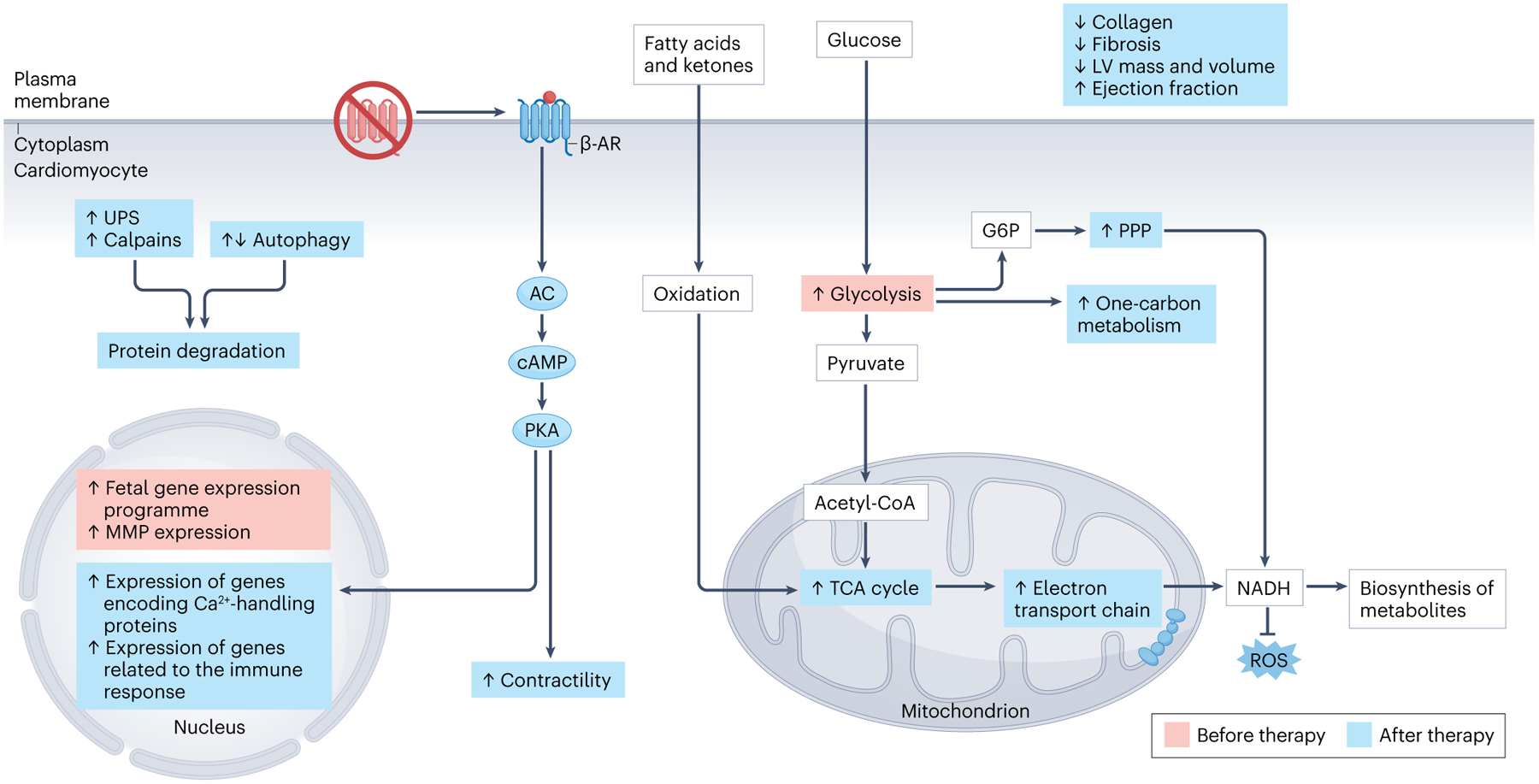
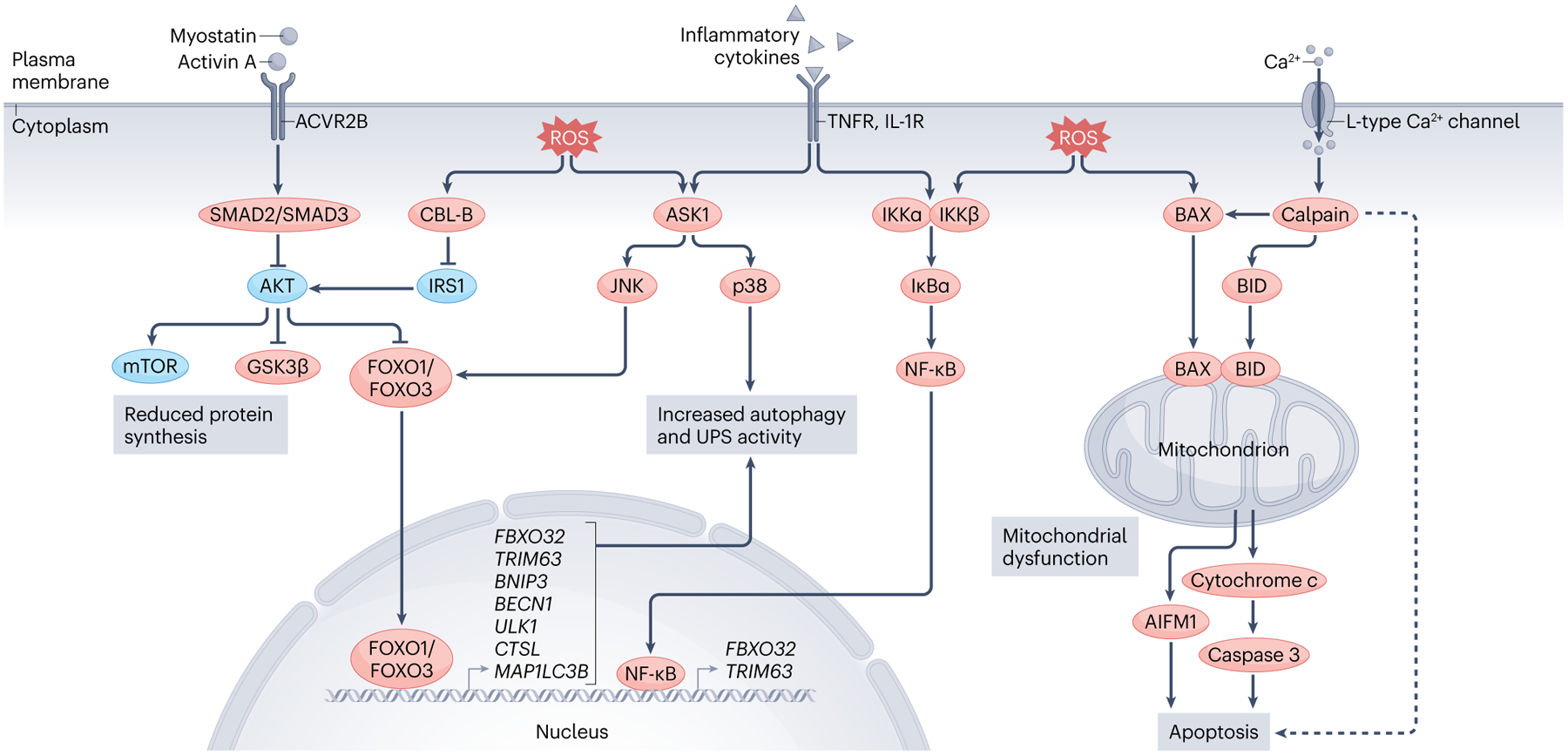
Similar articles
-
Mechanisms of physiological and pathological cardiac hypertrophy.Nat Rev Cardiol. 2018 Jul;15(7):387-407. doi: 10.1038/s41569-018-0007-y. Nat Rev Cardiol. 2018. PMID: 29674714 Review.
-
Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies.Pharmacol Ther. 2010 Oct;128(1):191-227. doi: 10.1016/j.pharmthera.2010.04.005. Epub 2010 May 12. Pharmacol Ther. 2010. PMID: 20438756 Review.
-
Eccentric and concentric cardiac hypertrophy induced by exercise training: microRNAs and molecular determinants.Braz J Med Biol Res. 2011 Sep;44(9):836-47. doi: 10.1590/s0100-879x2011007500112. Epub 2011 Sep 2. Braz J Med Biol Res. 2011. PMID: 21881810 Review.
-
Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling.J Am Heart Assoc. 2013 Apr 23;2(2):e000078. doi: 10.1161/JAHA.113.000078. J Am Heart Assoc. 2013. PMID: 23612897 Free PMC article.
-
The therapeutic potential of miRNAs regulated in settings of physiological cardiac hypertrophy.Future Med Chem. 2014 Feb;6(2):205-22. doi: 10.4155/fmc.13.196. Future Med Chem. 2014. PMID: 24467244 Review.
Cited by
-
Plasma and Myocardial miRNomes Similarities and Differences during Cardiac Remodelling and Reverse Remodelling in a Murine Model of Heart Failure with Preserved Ejection Fraction.Biomolecules. 2024 Jul 24;14(8):892. doi: 10.3390/biom14080892. Biomolecules. 2024. PMID: 39199280 Free PMC article.
-
Therapeutic potentials of allicin in cardiovascular disease: advances and future directions.Chin Med. 2024 Jul 2;19(1):93. doi: 10.1186/s13020-024-00936-8. Chin Med. 2024. PMID: 38956680 Free PMC article. Review.
-
Mitochondrial Melatonin: Beneficial Effects in Protecting against Heart Failure.Life (Basel). 2024 Jan 5;14(1):88. doi: 10.3390/life14010088. Life (Basel). 2024. PMID: 38255703 Free PMC article. Review.
-
Regression of postprandial cardiac hypertrophy in burmese pythons is mediated by FoxO1.Proc Natl Acad Sci U S A. 2024 Oct 8;121(41):e2408719121. doi: 10.1073/pnas.2408719121. Epub 2024 Oct 1. Proc Natl Acad Sci U S A. 2024. PMID: 39352930
-
A Conserved Mechanism of Cardiac Hypertrophy Regression through FoxO1.bioRxiv [Preprint]. 2024 Jan 28:2024.01.27.577585. doi: 10.1101/2024.01.27.577585. bioRxiv. 2024. Update in: Proc Natl Acad Sci U S A. 2024 Oct 8;121(41):e2408719121. doi: 10.1073/pnas.2408719121 PMID: 38328143 Free PMC article. Updated. Preprint.
References
-
- Levy D, Garrison RJ, Savage DD, Kannel WB & Castelli WP Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med 322, 1561–1566 (1990). - PubMed
-
- Izumi C et al. Effect of left ventricular reverse remodeling on long-term outcomes after aortic valve replacement. Am. J. Cardiol 124, 105–112 (2019). - PubMed
-
- Daubert MA et al. NT-proBNP goal achievement is associated with significant reverse remodeling and improved clinical outcomes in HFrEF. JACC Heart Fail. 7, 158–168 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
