

SARS-CoV-2 proteases Mpro and PLpro: Design of inhibitors with predicted high potency and low mammalian toxicity using artificial neural networks, ligand-protein docking, molecular dynamics simulations, and ADMET calculations
- PMID: 36586228
- PMCID: PMC9788855
- DOI: 10.1016/j.compbiomed.2022.106449
SARS-CoV-2 proteases Mpro and PLpro: Design of inhibitors with predicted high potency and low mammalian toxicity using artificial neural networks, ligand-protein docking, molecular dynamics simulations, and ADMET calculations
Abstract
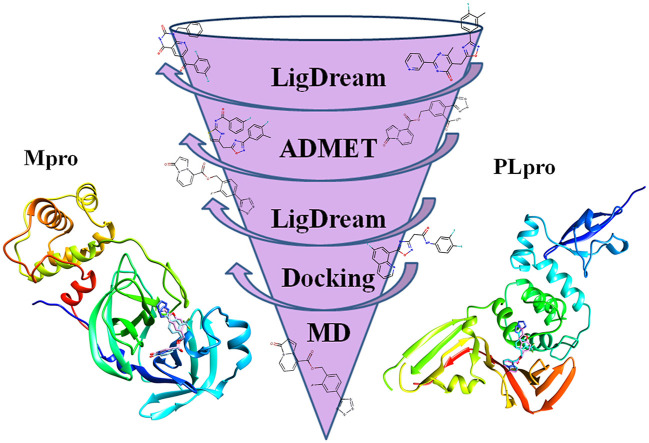
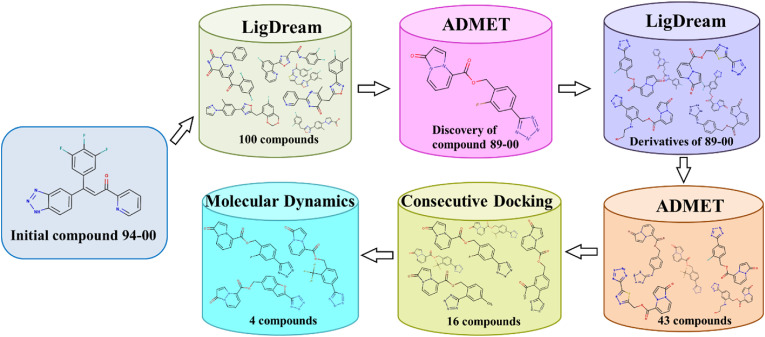
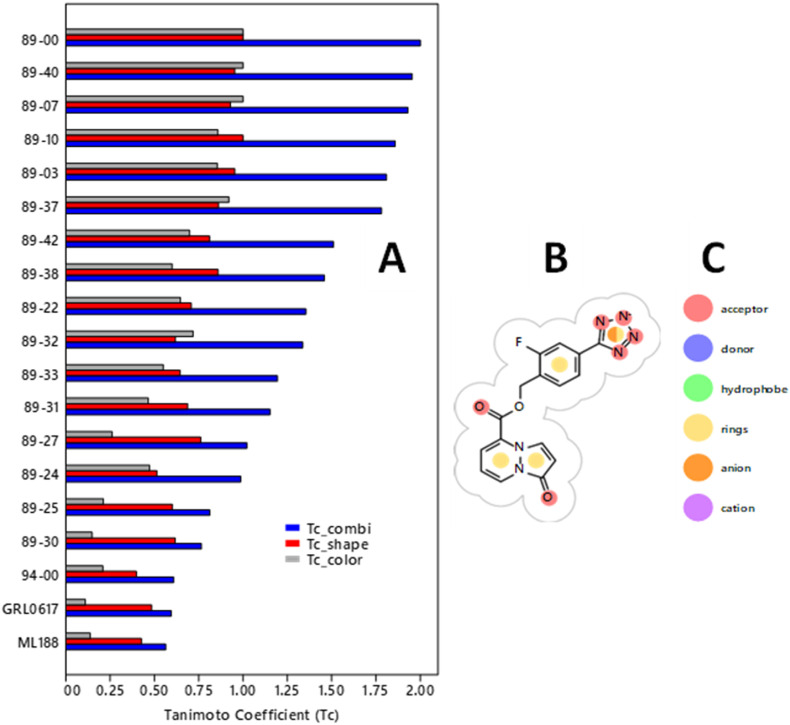
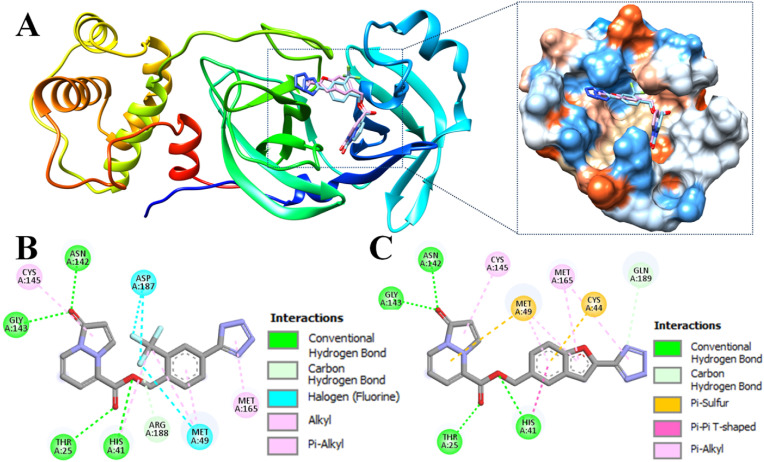
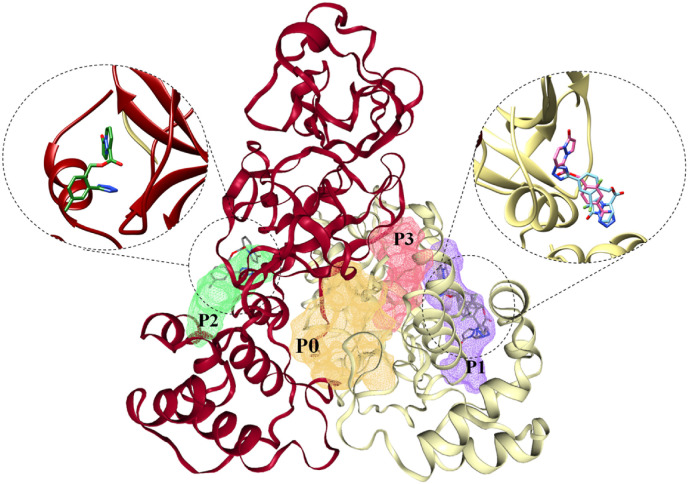
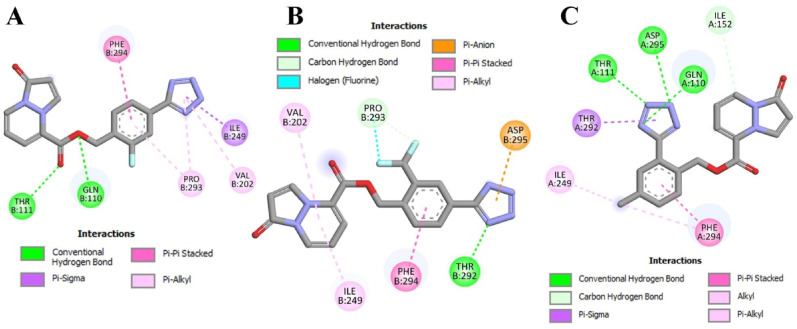
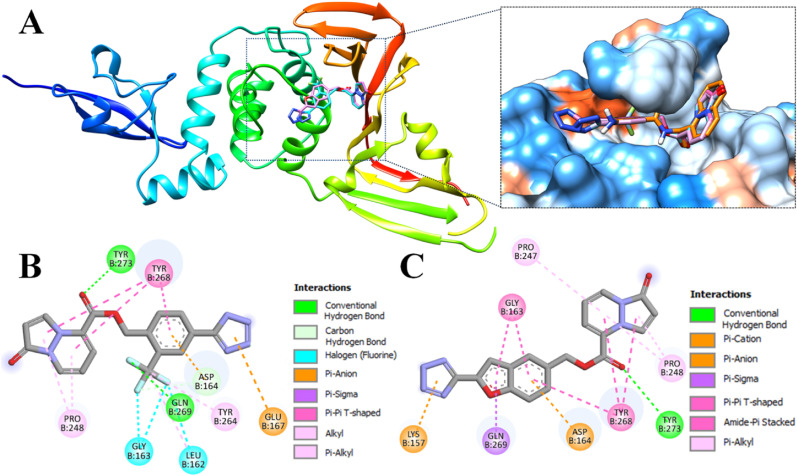
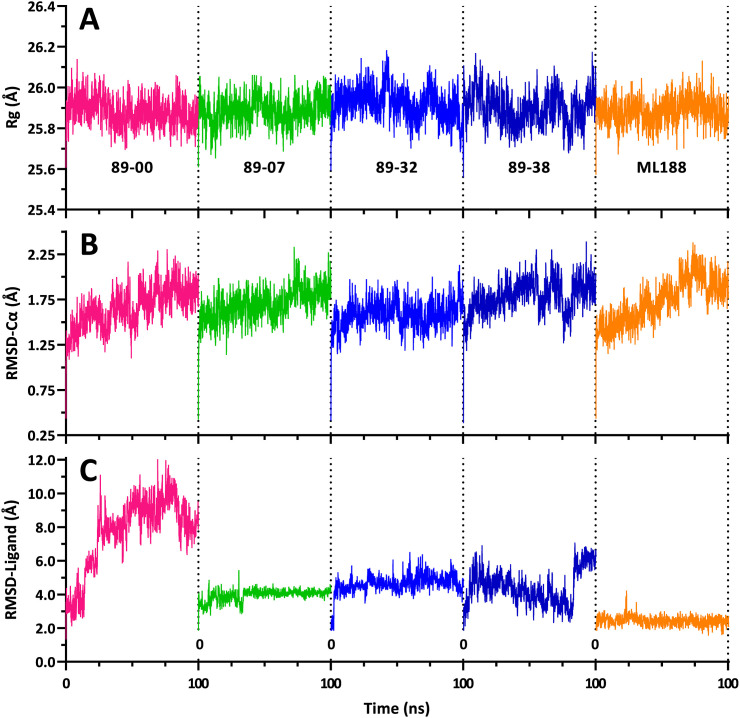
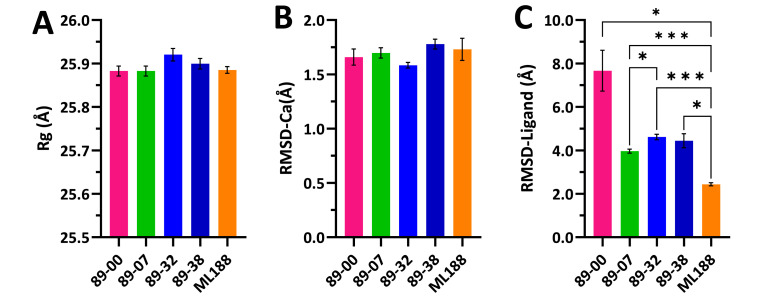
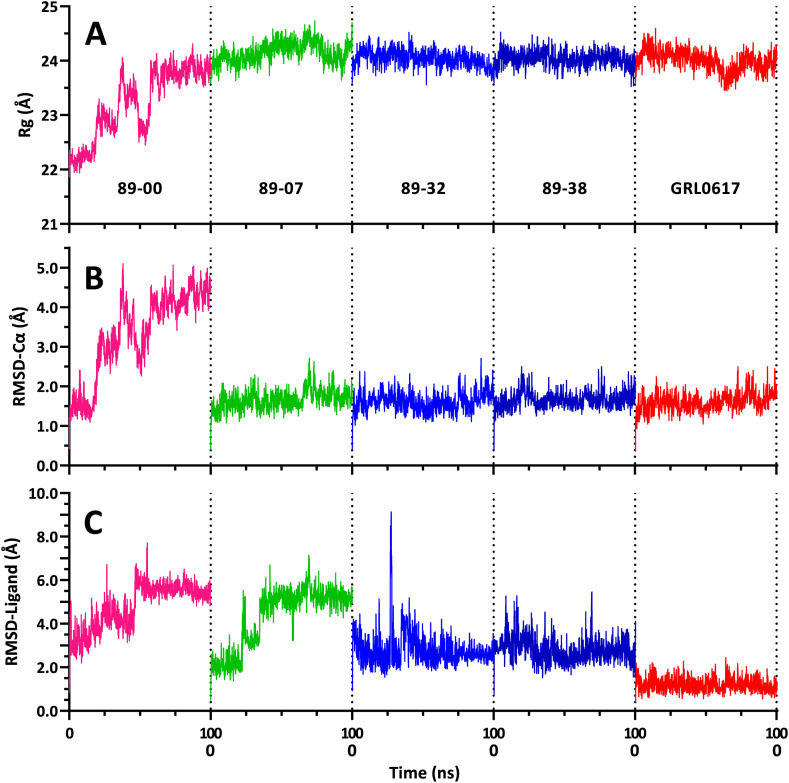
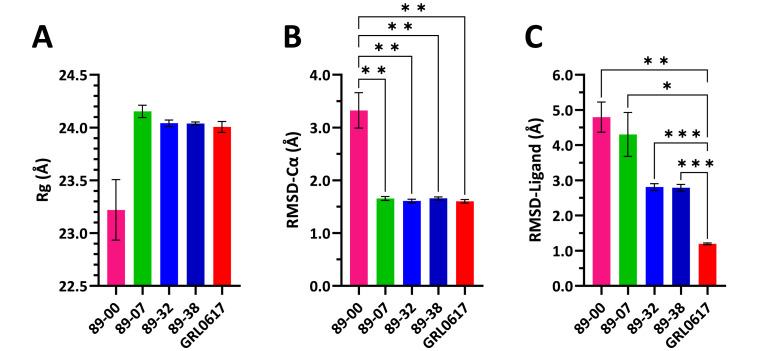
The main (Mpro) and papain-like (PLpro) proteases are highly conserved viral proteins essential for replication of the COVID-19 virus, SARS-COV-2. Therefore, a logical plan for producing new drugs against this pathogen is to discover inhibitors of these enzymes. Accordingly, the goal of the present work was to devise a computational approach to design, characterize, and select compounds predicted to be potent dual inhibitors - effective against both Mpro and PLpro. The first step employed LigDream, an artificial neural network, to create a virtual ligand library. Ligands with computed ADMET profiles indicating drug-like properties and low mammalian toxicity were selected for further study. Initial docking of these ligands into the active sites of Mpro and PLpro was done with GOLD, and the highest-scoring ligands were redocked with AutoDock Vina to determine binding free energies (ΔG). Compounds 89-00, 89-07, 89-32, and 89-38 exhibited favorable ΔG values for Mpro (-7.6 to -8.7 kcal/mol) and PLpro (-9.1 to -9.7 kcal/mol). Global docking of selected compounds with the Mpro dimer identified prospective allosteric inhibitors 89-00, 89-27, and 89-40 (ΔG -8.2 to -8.9 kcal/mol). Molecular dynamics simulations performed on Mpro and PLpro active site complexes with the four top-scoring ligands from Vina demonstrated that the most stable complexes were formed with compounds 89-32 and 89-38. Overall, the present computational strategy generated new compounds with predicted drug-like characteristics, low mammalian toxicity, and high inhibitory potencies against both target proteases to form stable complexes. Further preclinical studies will be required to validate the in silico findings before the lead compounds could be considered for clinical trials.
Keywords: ADMET; COVID-19; Heterocyclic compounds; In silico drug design; Molecular docking; Molecular dynamics simulation; Mpro/PLpro inhibitors; Nirmatrelvir; Pyrazolopyridazines; Tetrazoles.
Copyright © 2022 The Authors. Published by Elsevier Ltd.. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have influenced, or appear to have influenced, the work reported in this paper.
Figures
Similar articles
-
Computational Evidences of Phytochemical Mediated Disruption of PLpro Driven Replication of SARS-CoV-2: A Therapeutic Approach against COVID-19.Curr Pharm Biotechnol. 2021;22(10):1350-1359. doi: 10.2174/1389201021999201110204116. Curr Pharm Biotechnol. 2021. PMID: 33176643
-
Identification of potential plant-based inhibitor against viral proteases of SARS-CoV-2 through molecular docking, MM-PBSA binding energy calculations and molecular dynamics simulation.Mol Divers. 2021 Aug;25(3):1963-1977. doi: 10.1007/s11030-021-10211-9. Epub 2021 Apr 15. Mol Divers. 2021. PMID: 33856591 Free PMC article.
-
In silico screening of phytopolyphenolics for the identification of bioactive compounds as novel protease inhibitors effective against SARS-CoV-2.J Biomol Struct Dyn. 2022;40(20):10437-10453. doi: 10.1080/07391102.2021.1944909. Epub 2021 Jun 28. J Biomol Struct Dyn. 2022. PMID: 34182889
-
Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs.Precis Clin Med. 2021 Jan 18;4(1):1-16. doi: 10.1093/pcmedi/pbab001. eCollection 2021 Mar. Precis Clin Med. 2021. PMID: 33842834 Free PMC article. Review.
-
Covalent small-molecule inhibitors of SARS-CoV-2 Mpro: Insights into their design, classification, biological activity, and binding interactions.Eur J Med Chem. 2024 Nov 5;277:116704. doi: 10.1016/j.ejmech.2024.116704. Epub 2024 Aug 8. Eur J Med Chem. 2024. PMID: 39121741 Review.
Cited by
-
Covalent Inhibitors from Saudi Medicinal Plants Target RNA-Dependent RNA Polymerase (RdRp) of SARS-CoV-2.Viruses. 2023 Oct 30;15(11):2175. doi: 10.3390/v15112175. Viruses. 2023. PMID: 38005857 Free PMC article.
-
Identification of Phytochemicals from Arabian Peninsula Medicinal Plants as Strong Binders to SARS-CoV-2 Proteases (3CLPro and PLPro) by Molecular Docking and Dynamic Simulation Studies.Molecules. 2024 Feb 25;29(5):998. doi: 10.3390/molecules29050998. Molecules. 2024. PMID: 38474509 Free PMC article.
-
The impact of SARS-CoV-2 3CL protease mutations on nirmatrelvir inhibitory efficiency. Computational insights into potential resistance mechanisms.Chem Sci. 2023 Feb 14;14(10):2686-2697. doi: 10.1039/d2sc06584c. eCollection 2023 Mar 8. Chem Sci. 2023. PMID: 36908962 Free PMC article.
-
A combination of virtual screening, molecular dynamics simulation, MM/PBSA, ADMET, and DFT calculations to identify a potential DPP4 inhibitor.Sci Rep. 2024 Apr 2;14(1):7749. doi: 10.1038/s41598-024-58485-x. Sci Rep. 2024. PMID: 38565703 Free PMC article.
-
Main and papain-like proteases as prospective targets for pharmacological treatment of coronavirus SARS-CoV-2.RSC Adv. 2023 Dec 6;13(50):35500-35524. doi: 10.1039/d3ra06479d. eCollection 2023 Nov 30. RSC Adv. 2023. PMID: 38077980 Free PMC article. Review.
References
-
- World Health Organization 2022. https://www.who.int/emergencies/diseases/novel-coronavirus-2019
-
- World Health Organization 2022. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
