

The PI3K/Akt Pathway in Meta-Inflammation
- PMID: 36499659
- PMCID: PMC9740745
- DOI: 10.3390/ijms232315330
The PI3K/Akt Pathway in Meta-Inflammation
Abstract
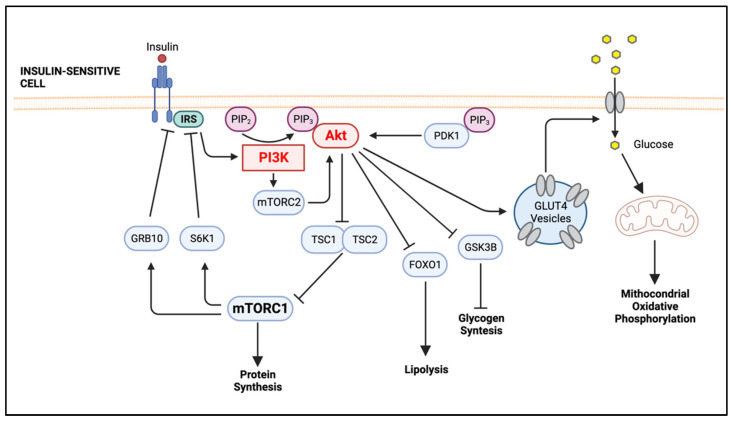
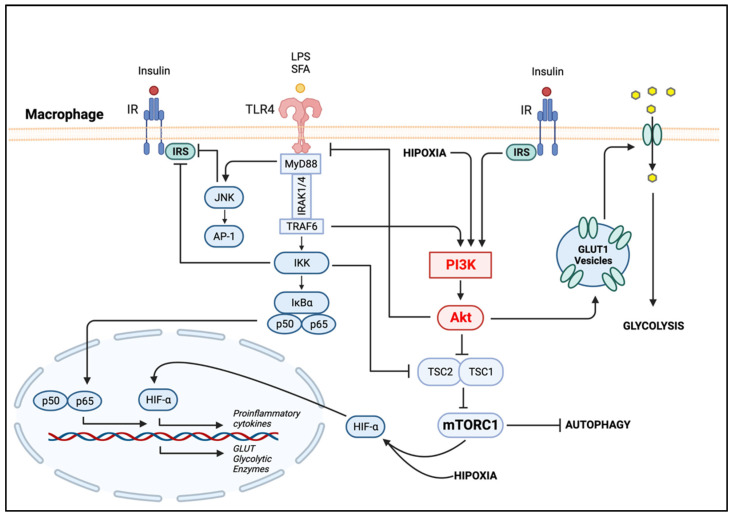
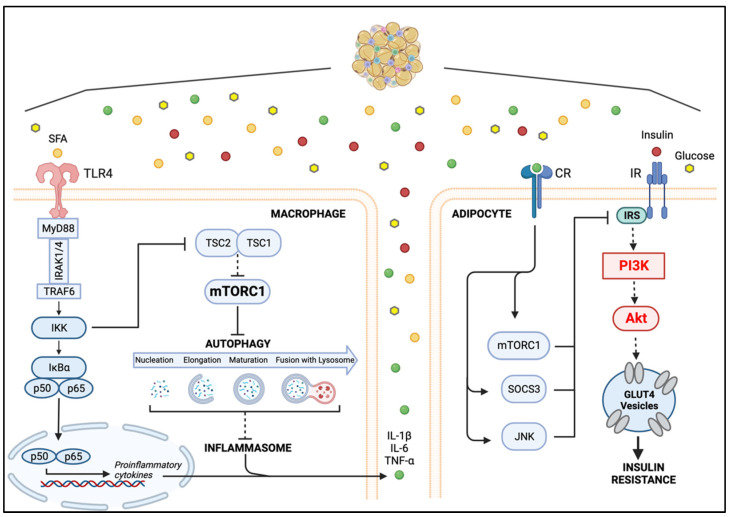
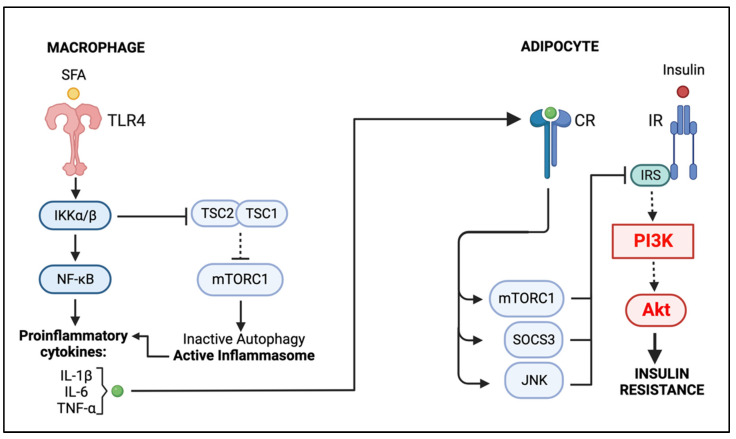
Obesity is a global epidemic representing a serious public health burden as it is a major risk factor for the development of cardiovascular disease, stroke and all-cause mortality. Chronic low-grade systemic inflammation, also known as meta-inflammation, is thought to underly obesity's negative health consequences, which include insulin resistance and the development of type 2 diabetes. Meta-inflammation is characterized by the accumulation of immune cells in adipose tissue, a deregulation in the synthesis and release of adipokines and a pronounced increase in the production of proinflammatory factors. In this state, the infiltration of macrophages and their metabolic activation contributes to complex paracrine and autocrine signaling, which sustains a proinflammatory microenvironment. A key signaling pathway mediating the response of macrophages and adipocytes to a microenvironment of excessive nutrients is the phosphoinositide 3-kinase (PI3K)/Akt pathway. This multifaceted network not only transduces metabolic information but also regulates macrophages' intracellular changes, which are responsible for their phenotypic switch towards a more proinflammatory state. In the present review, we discuss how the crosstalk between macrophages and adipocytes contributes to meta-inflammation and provide an overview on the involvement of the PI3K/Akt signaling pathway, and how its impairment contributes to the development of insulin resistance.
Keywords: Akt; PI3K; adipocytes; macrophages; meta-inflammation; obesity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance.Nutrients. 2020 May 3;12(5):1305. doi: 10.3390/nu12051305. Nutrients. 2020. PMID: 32375231 Free PMC article. Review.
-
Adrenomedullin ameliorates palmitic acid-induced insulin resistance through PI3K/Akt pathway in adipocytes.Acta Diabetol. 2022 May;59(5):661-673. doi: 10.1007/s00592-021-01840-5. Epub 2022 Jan 3. Acta Diabetol. 2022. PMID: 34978596
-
The PI3K/AKT pathway in obesity and type 2 diabetes.Int J Biol Sci. 2018 Aug 6;14(11):1483-1496. doi: 10.7150/ijbs.27173. eCollection 2018. Int J Biol Sci. 2018. PMID: 30263000 Free PMC article. Review.
-
SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes.Diabetologia. 2017 Jul;60(7):1304-1313. doi: 10.1007/s00125-017-4261-z. Epub 2017 Apr 5. Diabetologia. 2017. PMID: 28382382 Free PMC article.
-
Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance.Mol Cell Biochem. 2017 Feb;426(1-2):27-45. doi: 10.1007/s11010-016-2878-8. Epub 2016 Nov 21. Mol Cell Biochem. 2017. PMID: 27868170 Free PMC article. Review.
Cited by
-
CXCR4 regulates macrophage M1 polarization by altering glycolysis to promote prostate fibrosis.Cell Commun Signal. 2024 Sep 26;22(1):456. doi: 10.1186/s12964-024-01828-y. Cell Commun Signal. 2024. PMID: 39327570 Free PMC article.
-
Intestinal Motility Dysfunction in Goto-Kakizaki Rats: Role of the Myenteric Plexus.Cells. 2024 Sep 28;13(19):1626. doi: 10.3390/cells13191626. Cells. 2024. PMID: 39404390 Free PMC article.
-
Syringin from Tinospora crispa downregulates pro-inflammatory mediator production through MyD88-dependent pathways in lipopolysaccharide (LPS)-induced U937 macrophages.Mol Biol Rep. 2024 Jul 11;51(1):789. doi: 10.1007/s11033-024-09722-z. Mol Biol Rep. 2024. PMID: 38990383
-
The effects of exercise on microRNA expression profiling in adipose tissue macrophages of mice.Front Immunol. 2024 Aug 19;15:1412621. doi: 10.3389/fimmu.2024.1412621. eCollection 2024. Front Immunol. 2024. PMID: 39224599 Free PMC article.
-
Mitigation of acetaminophen-induced liver toxicity by the novel phosphatidylinositol 3-kinase inhibitor alpelisib.Front Pharmacol. 2023 Aug 7;14:1212771. doi: 10.3389/fphar.2023.1212771. eCollection 2023. Front Pharmacol. 2023. PMID: 37608890 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
