

Insights into the structural properties of SARS-CoV-2 main protease
- PMID: 36466947
- PMCID: PMC9700396
- DOI: 10.1016/j.crstbi.2022.11.001
Insights into the structural properties of SARS-CoV-2 main protease
Abstract
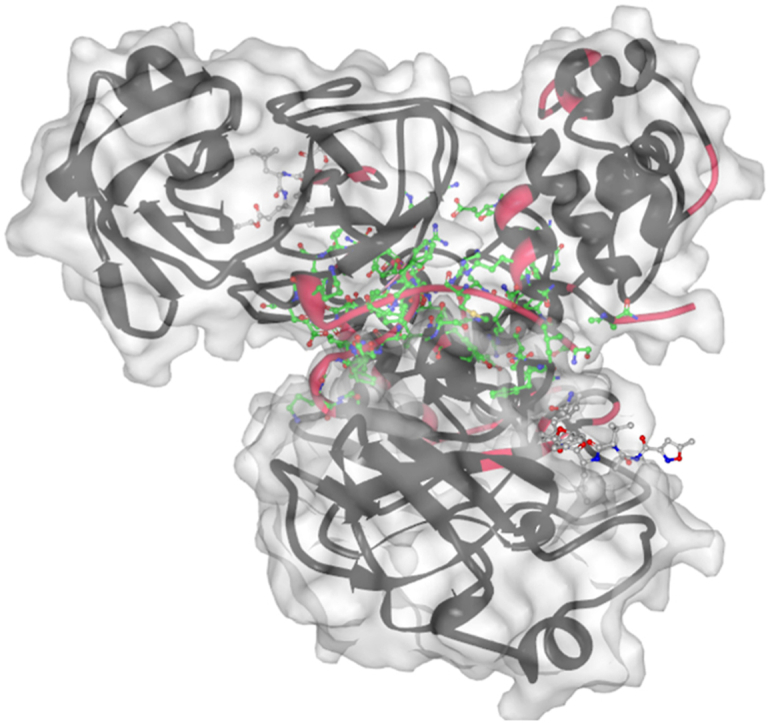
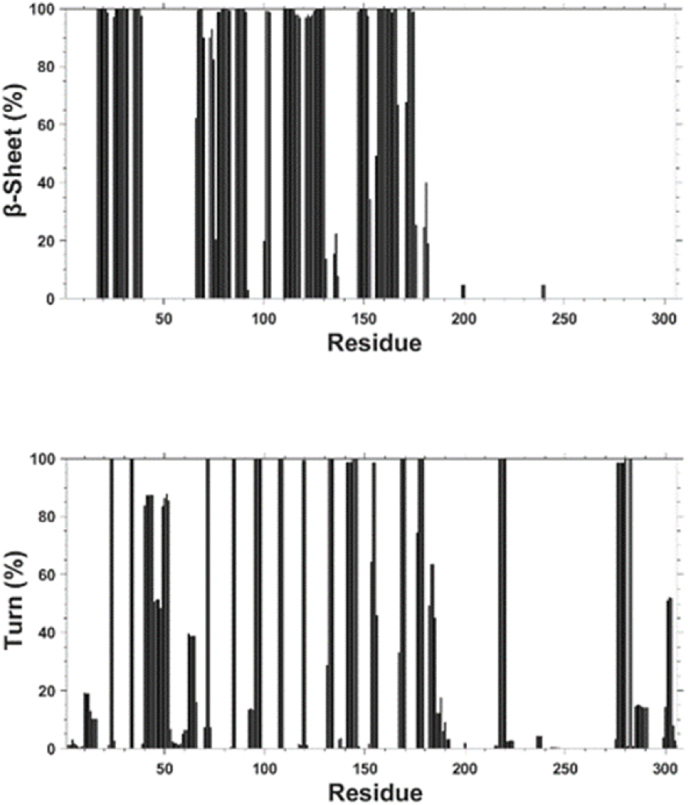
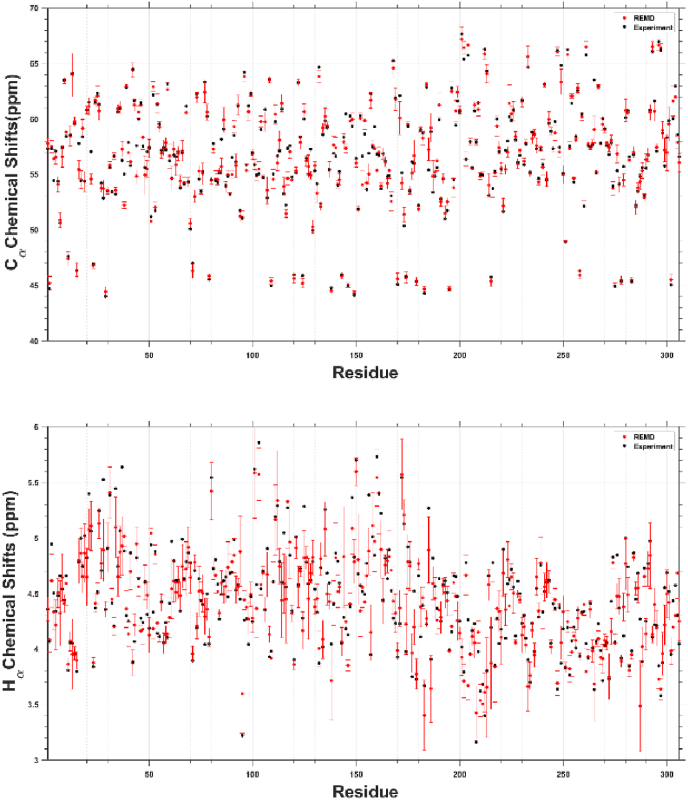
SARS-CoV-2 is the infectious agent responsible for the coronavirus disease since 2019, which is the viral pneumonia pandemic worldwide. The structural knowledge on SARS-CoV-2 is rather limited. These limitations are also applicable to one of the most attractive drug targets of SARS-CoV-2 proteins - namely, main protease Mpro, also known as 3C-like protease (3CLpro). This protein is crucial for the processing of the viral polyproteins and plays crucial roles in interfering viral replication and transcription. In fact, although the crystal structure of this protein with an inhibitor was solved, Mpro conformational dynamics in aqueous solution is usually studied by molecular dynamics simulations without special sampling techniques. We conducted replica exchange molecular dynamics simulations on Mpro in water and report the dynamic structures of Mpro in an aqueous environment including root mean square fluctuations, secondary structure properties, radius of gyration, and end-to-end distances, chemical shift values, intrinsic disorder characteristics of Mpro and its active sites with a set of computational tools. The active sites we found coincide with the currently known sites and include a new interface for interaction with a protein partner.
Keywords: Dynamics; Replica exchange MD simulations; SARS-CoV-2 main protease.
© 2022 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures




Similar articles
-
Allosteric Regulation of 3CL Protease of SARS-CoV-2 and SARS-CoV Observed in the Crystal Structure Ensemble.J Mol Biol. 2021 Dec 3;433(24):167324. doi: 10.1016/j.jmb.2021.167324. Epub 2021 Oct 27. J Mol Biol. 2021. PMID: 34717972 Free PMC article.
-
Identification of a novel inhibitor of SARS-CoV-2 3CL-PRO through virtual screening and molecular dynamics simulation.PeerJ. 2021 Apr 13;9:e11261. doi: 10.7717/peerj.11261. eCollection 2021. PeerJ. 2021. PMID: 33954055 Free PMC article.
-
Identification of lead compounds from large natural product library targeting 3C-like protease of SARS-CoV-2 using E-pharmacophore modelling, QSAR and molecular dynamics simulation.In Silico Pharmacol. 2021 Aug 7;9(1):49. doi: 10.1007/s40203-021-00109-7. eCollection 2021. In Silico Pharmacol. 2021. PMID: 34395160 Free PMC article.
-
Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site.Viruses. 2020 Aug 26;12(9):942. doi: 10.3390/v12090942. Viruses. 2020. PMID: 32859008 Free PMC article.
-
Characterization of host substrates of SARS-CoV-2 main protease.Front Microbiol. 2023 Aug 21;14:1251705. doi: 10.3389/fmicb.2023.1251705. eCollection 2023. Front Microbiol. 2023. PMID: 37670988 Free PMC article. Review.
Cited by
-
Structural Properties of Rat Intestinal Fatty Acid-Binding Protein with its Dynamics: Insights into Intrinsic Disorder.Protein Pept Lett. 2024;31(6):458-468. doi: 10.2174/0109298665313811240530055004. Protein Pept Lett. 2024. PMID: 38910419
References
-
- Akaji K., et al. Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J. Med. Chem. 2011;54:7962–7973. - PubMed
-
- Akbayrak I.Y., Caglayan S.I., Durdagi S., Kurgan L., Uversky V.N., Ulver B., Dervisoglu H., Haklidir M., Hasekioglu O., Coskuner-Weber O. Structures of MERS-CoV macro domain in aqueous solution with dynamics: impacts of parallel tempering simulation techniques and CHARMM36m and AMBER99SB force field parameters. Proteins: Struct., Funct., Bioinf. 2021;89(10):1289–1299. doi: 10.1002/prot.26150. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
