

Cross-regulome profiling of RNA polymerases highlights the regulatory role of polymerase III on mRNA transcription by maintaining local chromatin architecture
- PMID: 36443871
- PMCID: PMC9703767
- DOI: 10.1186/s13059-022-02812-w
Cross-regulome profiling of RNA polymerases highlights the regulatory role of polymerase III on mRNA transcription by maintaining local chromatin architecture
Abstract
Background: Mammalian cells have three types of RNA polymerases (Pols), Pol I, II, and III. However, the extent to which these polymerases are cross-regulated and the underlying mechanisms remain unclear.
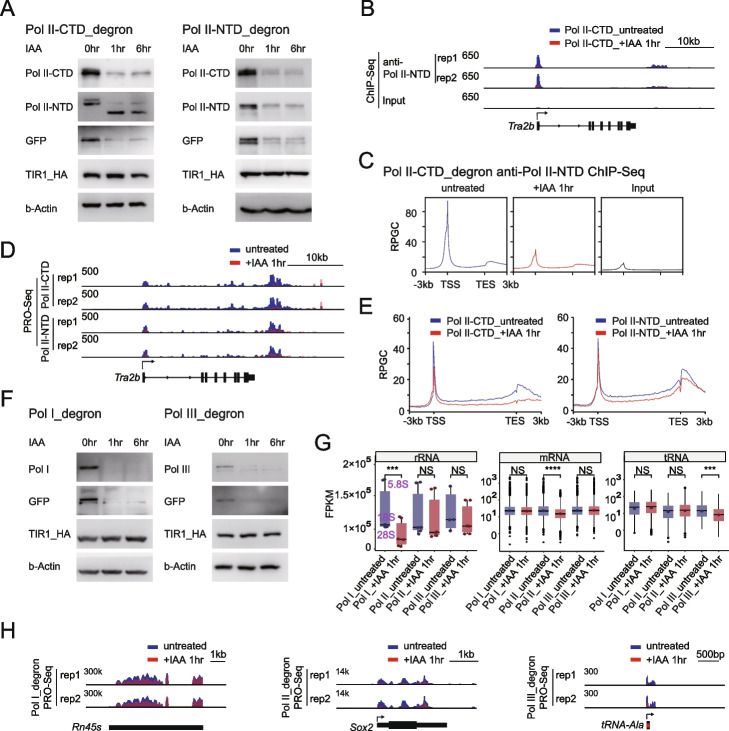
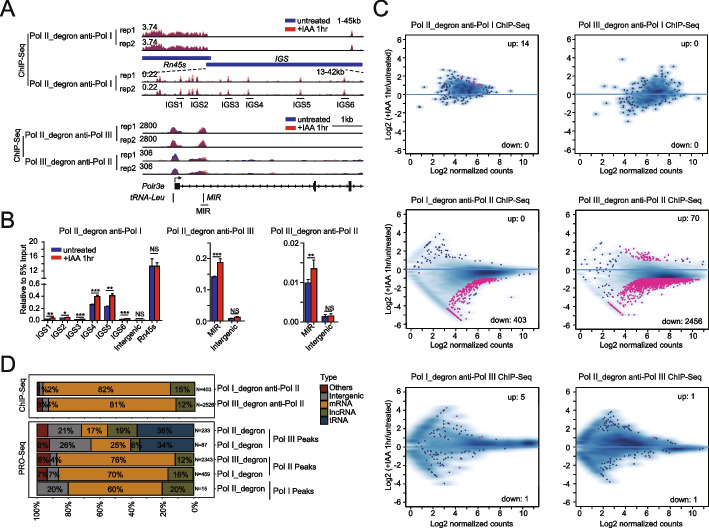
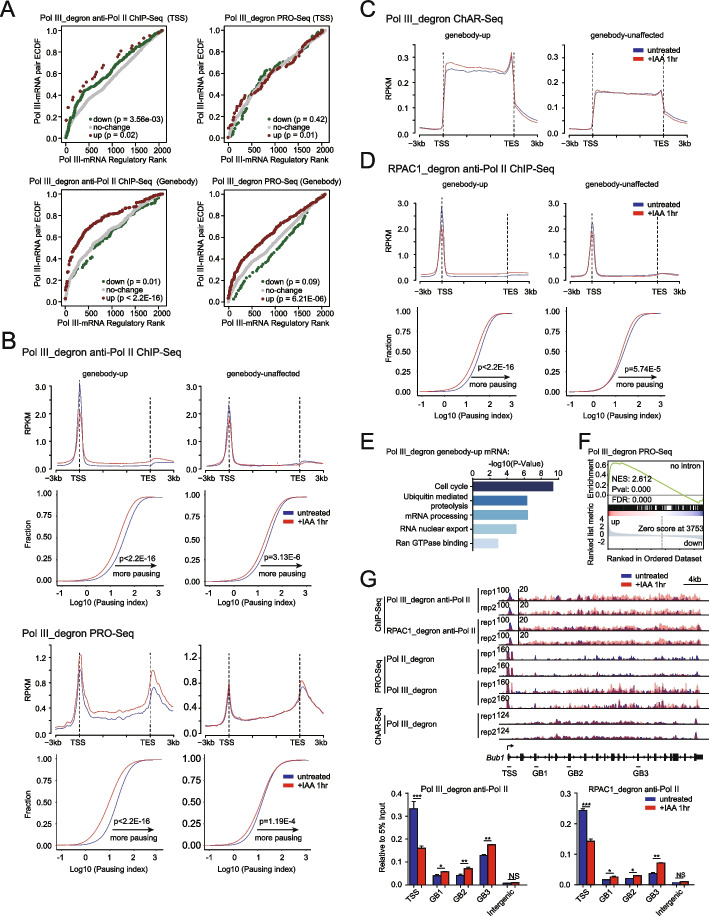
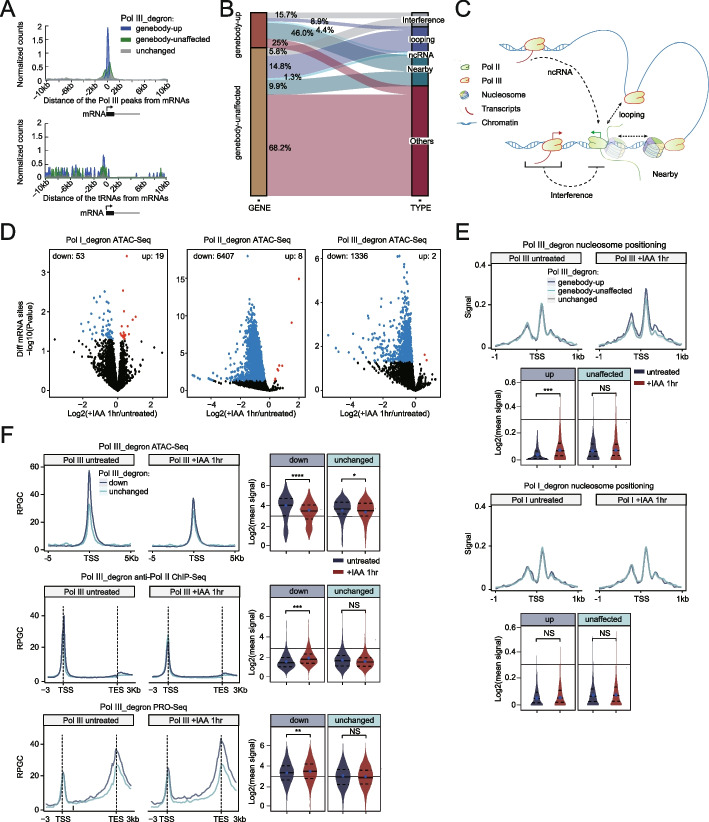
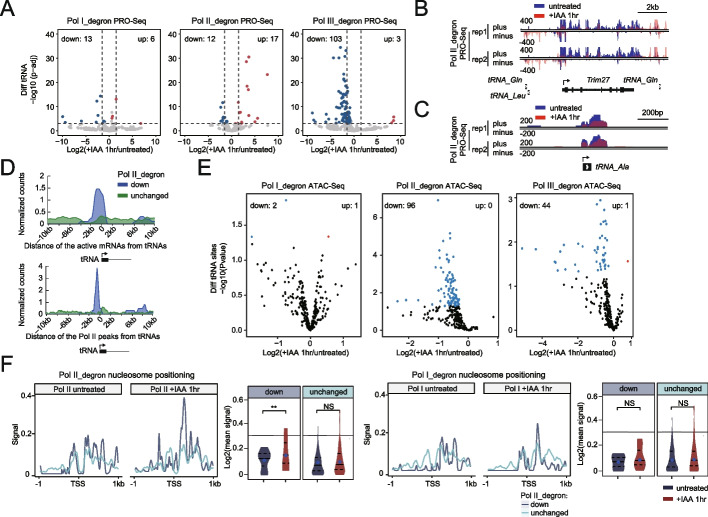
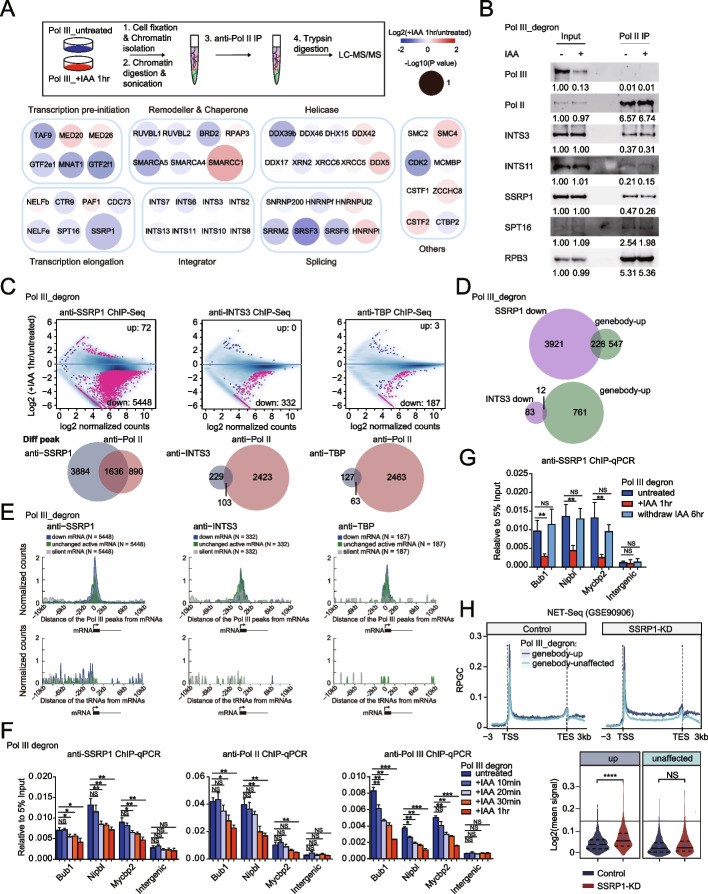
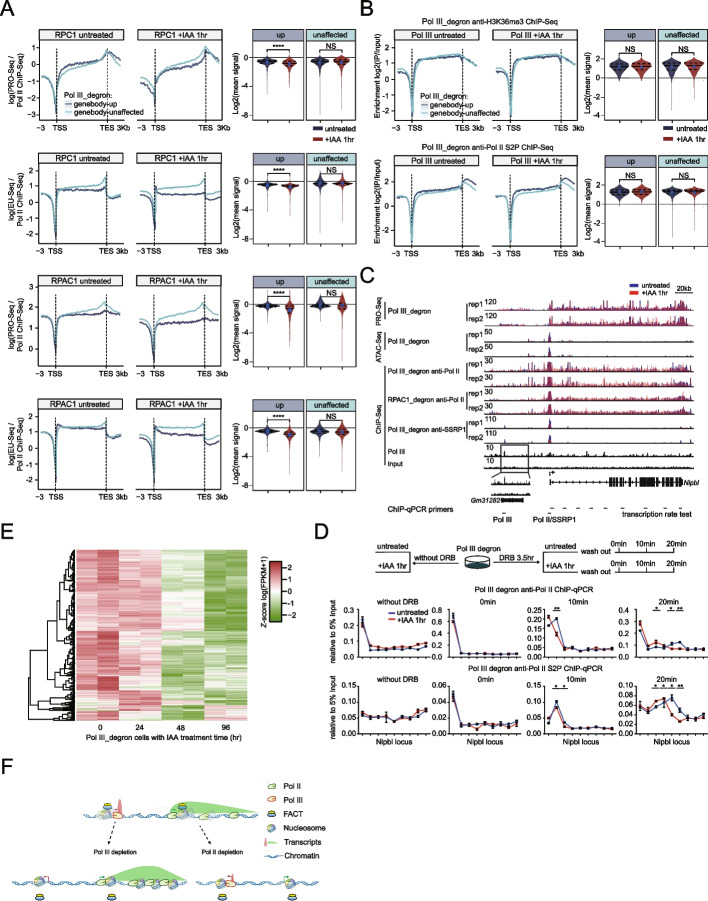
Results: We employ genome-wide profiling after acute depletion of Pol I, Pol II, or Pol III to assess cross-regulatory effects between these Pols. We find that these enzymes mainly affect the transcription of their own target genes, while certain genes are transcribed by the other polymerases. Importantly, the most active type of crosstalk is exemplified by the fact that Pol III depletion affects Pol II transcription. Pol II genes with transcription changes upon Pol III depletion are enriched in diverse cellular functions, and Pol III binding sites are found near their promoters. However, these Pol III binding sites do not correspond to transfer RNAs. Moreover, we demonstrate that Pol III regulates Pol II transcription and chromatin binding of the facilitates chromatin transcription (FACT) complex to alter local chromatin structures, which in turn affects the Pol II transcription rate.
Conclusions: Our results support a model suggesting that RNA polymerases show cross-regulatory effects: Pol III affects local chromatin structures and the FACT-Pol II axis to regulate the Pol II transcription rate at certain gene loci. This study provides a new perspective for understanding the dysregulation of Pol III in various tissues affected by developmental diseases.
Keywords: FACT complex; Local chromatin structure; RNA polymerases II and III; Transcription rate.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Genome-wide analyses of chromatin interactions after the loss of Pol I, Pol II, and Pol III.Genome Biol. 2020 Jul 2;21(1):158. doi: 10.1186/s13059-020-02067-3. Genome Biol. 2020. PMID: 32616013 Free PMC article.
-
FACT facilitates chromatin transcription by RNA polymerases I and III.EMBO J. 2009 Apr 8;28(7):854-65. doi: 10.1038/emboj.2009.33. Epub 2009 Feb 12. EMBO J. 2009. PMID: 19214185 Free PMC article.
-
RNA polymerases IV and V influence the 3' boundaries of Polymerase II transcription units in Arabidopsis.RNA Biol. 2018 Feb 1;15(2):269-279. doi: 10.1080/15476286.2017.1409930. Epub 2017 Dec 21. RNA Biol. 2018. PMID: 29199514 Free PMC article.
-
Regulatory networking of the three RNA polymerases helps the eukaryotic cells cope with environmental stress.Curr Genet. 2021 Aug;67(4):595-603. doi: 10.1007/s00294-021-01179-y. Epub 2021 Mar 28. Curr Genet. 2021. PMID: 33778898 Review.
-
Structural insights into nuclear transcription by eukaryotic DNA-dependent RNA polymerases.Nat Rev Mol Cell Biol. 2022 Sep;23(9):603-622. doi: 10.1038/s41580-022-00476-9. Epub 2022 May 3. Nat Rev Mol Cell Biol. 2022. PMID: 35505252 Review.
Cited by
-
Biallelic variants in GTF3C5, a regulator of RNA polymerase III-mediated transcription, cause a multisystem developmental disorder.Hum Genet. 2024 Mar;143(3):437-453. doi: 10.1007/s00439-024-02656-3. Epub 2024 Mar 23. Hum Genet. 2024. PMID: 38520561
-
Never a dull enzyme, RNA polymerase II.Transcription. 2023 Nov;14(1-2):49-67. doi: 10.1080/21541264.2023.2208023. Epub 2023 May 2. Transcription. 2023. PMID: 37132022 Free PMC article. Review.
-
Evidence of RNA polymerase III recruitment and transcription at protein-coding gene promoters.Mol Cell. 2024 Nov 7;84(21):4111-4124.e5. doi: 10.1016/j.molcel.2024.09.019. Epub 2024 Oct 10. Mol Cell. 2024. PMID: 39393362
-
The Pol III transcriptome: Basic features, recurrent patterns, and emerging roles in cancer.Wiley Interdiscip Rev RNA. 2023 Sep-Oct;14(5):e1782. doi: 10.1002/wrna.1782. Epub 2023 Feb 8. Wiley Interdiscip Rev RNA. 2023. PMID: 36754845 Free PMC article. Review.
-
Non-canonical functions of enhancers: regulation of RNA polymerase III transcription, DNA replication, and V(D)J recombination.Trends Genet. 2024 Jun;40(6):471-479. doi: 10.1016/j.tig.2024.04.001. Epub 2024 Apr 19. Trends Genet. 2024. PMID: 38643034
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
