

Impact of Siponimod on Enteric and Central Nervous System Pathology in Late-Stage Experimental Autoimmune Encephalomyelitis
- PMID: 36430692
- PMCID: PMC9695324
- DOI: 10.3390/ijms232214209
Impact of Siponimod on Enteric and Central Nervous System Pathology in Late-Stage Experimental Autoimmune Encephalomyelitis
Abstract
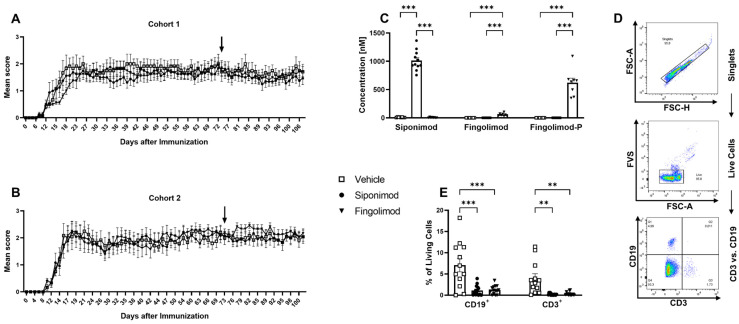
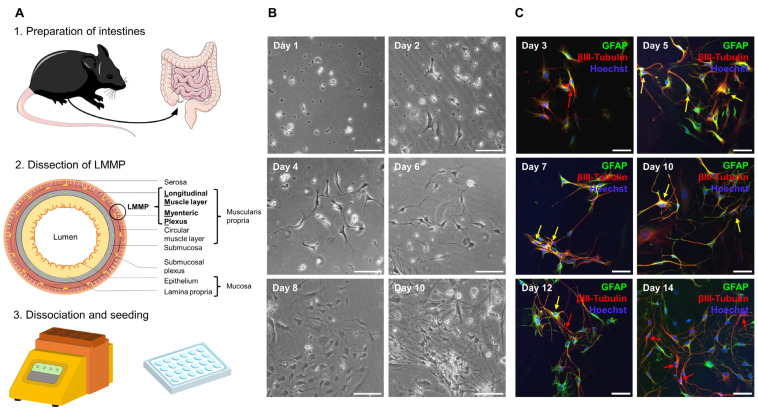
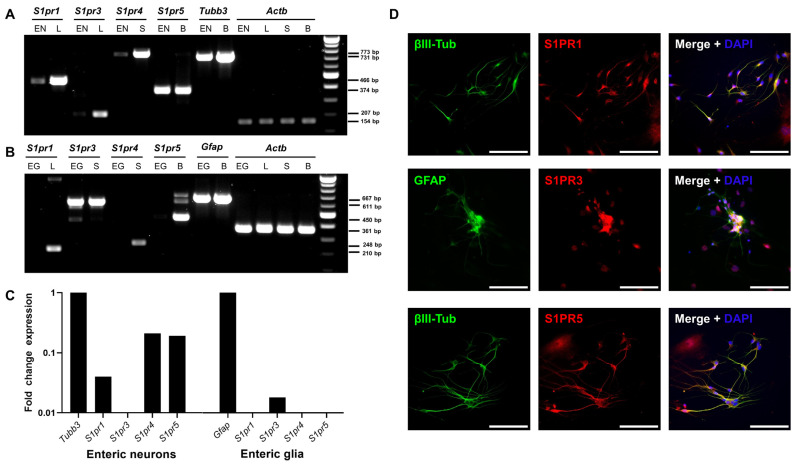
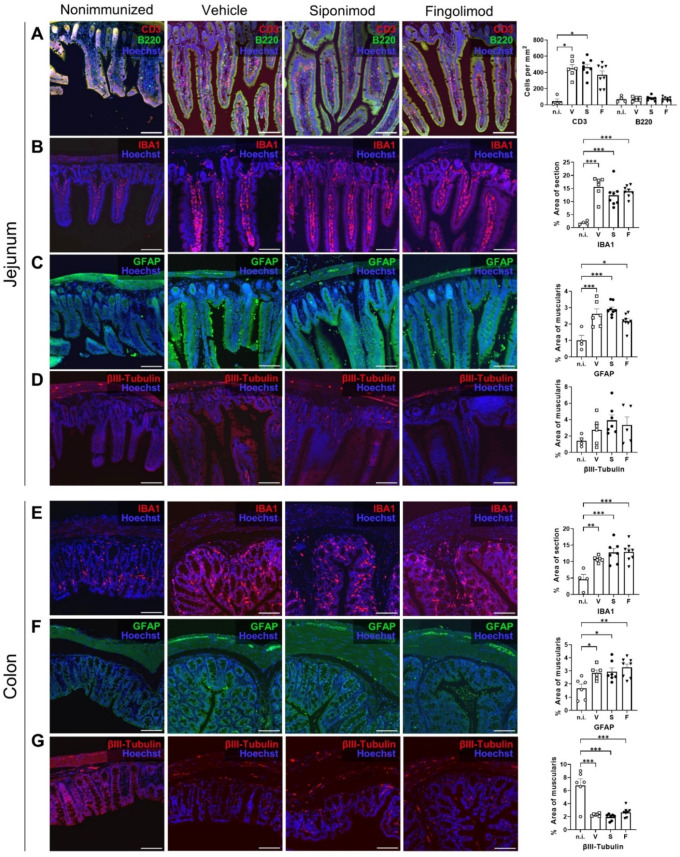
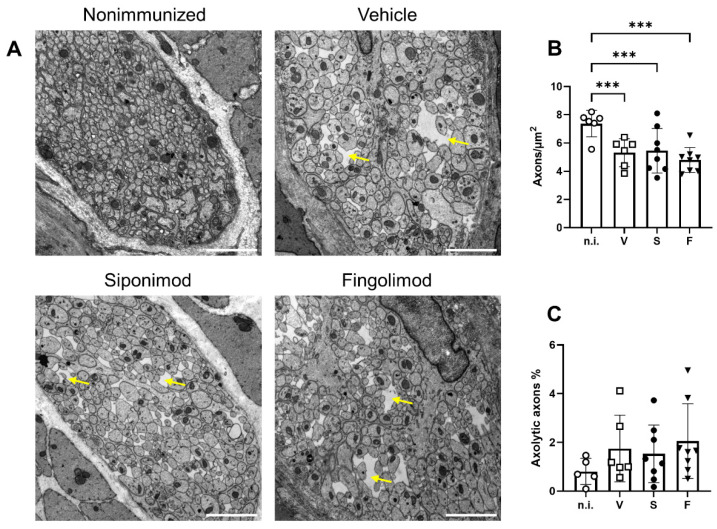
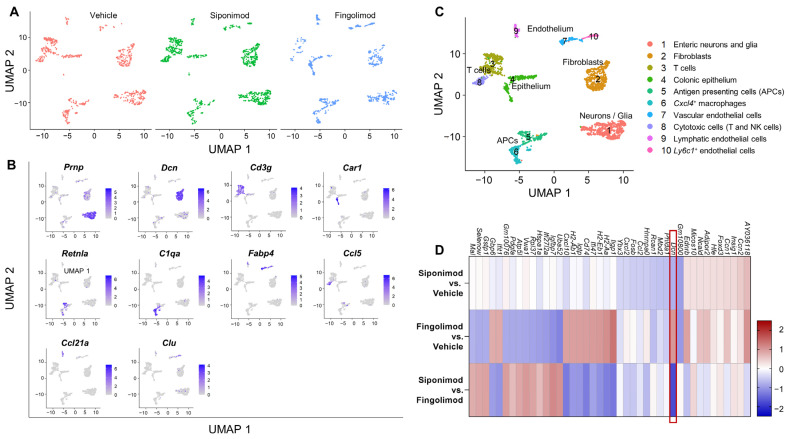
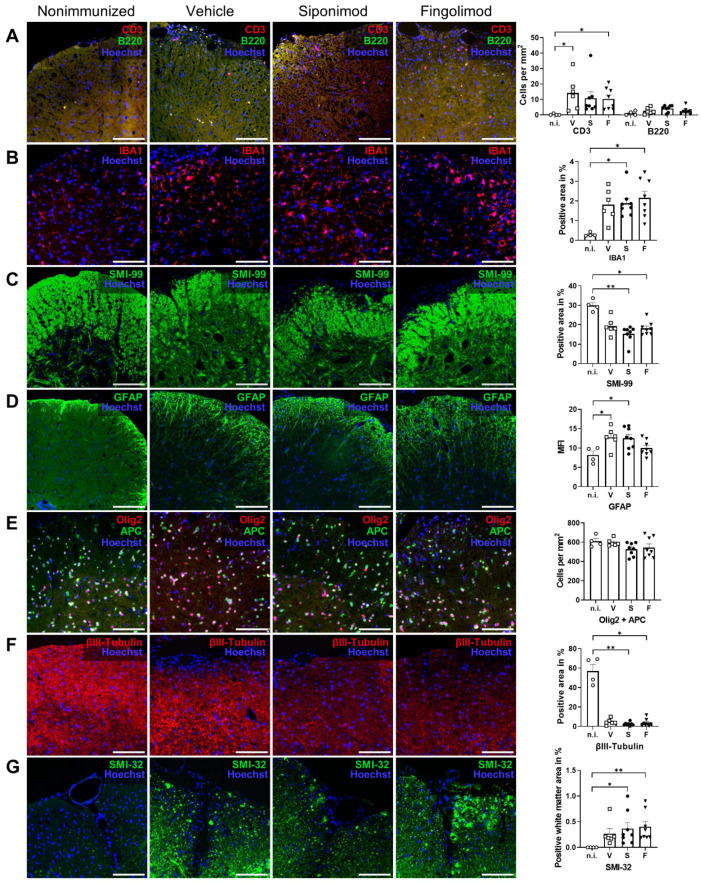
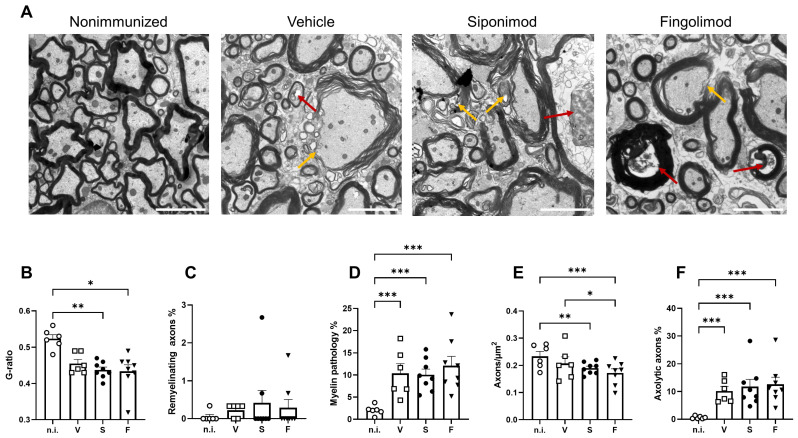
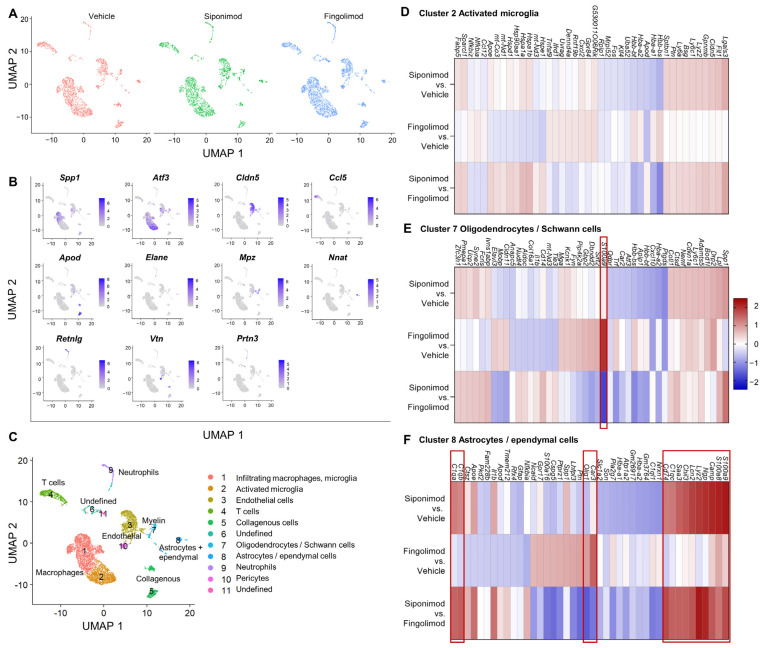
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS). Although immune modulation and suppression are effective during relapsing-remitting MS, secondary progressive MS (SPMS) requires neuroregenerative therapeutic options that act on the CNS. The sphingosine-1-phosphate receptor modulator siponimod is the only approved drug for SPMS. In the pivotal trial, siponimod reduced disease progression and brain atrophy compared with placebo. The enteric nervous system (ENS) was recently identified as an additional autoimmune target in MS. We investigated the effects of siponimod on the ENS and CNS in the experimental autoimmune encephalomyelitis model of MS. Mice with late-stage disease were treated with siponimod, fingolimod, or sham. The clinical disease was monitored daily, and treatment success was verified using mass spectrometry and flow cytometry, which revealed peripheral lymphopenia in siponimod- and fingolimod-treated mice. We evaluated the mRNA expression, ultrastructure, and histopathology of the ENS and CNS. Single-cell RNA sequencing revealed an upregulation of proinflammatory genes in spinal cord astrocytes and ependymal cells in siponimod-treated mice. However, differences in CNS and ENS histopathology and ultrastructural pathology between the treatment groups were absent. Thus, our data suggest that siponimod and fingolimod act on the peripheral immune system and do not have pronounced direct neuroprotective effects.
Keywords: central nervous system; enteric nervous system; experimental autoimmune encephalomyelitis; fingolimod; multiple sclerosis; siponimod.
Conflict of interest statement
A.W., M.E., P.K., A.E., C.K., and J.W. declare no conflicts of interest. M.B. is a Novartis employee and declares no further conflicts of interests. S.K. reports grants from the Deutsche Forschungsgemeinschaft (DFG), Novartis, F. Hoffmann-La Roche, and Sanofi; and speaker fees and consultancy honoraria from Novartis, F. Hoffmann-La Roche, Sanofi, and Teva (outside the submitted work). The funders had no role in the design of the study; in the interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
Similar articles
-
Siponimod treatment response shows partial BDNF dependency in multiple sclerosis models.Sci Rep. 2024 Aug 1;14(1):17823. doi: 10.1038/s41598-024-68715-x. Sci Rep. 2024. PMID: 39090252 Free PMC article.
-
Morpholino Analogues of Fingolimod as Novel and Selective S1P1 Ligands with In Vivo Efficacy in a Mouse Model of Experimental Antigen-Induced Encephalomyelitis.Int J Mol Sci. 2020 Sep 4;21(18):6463. doi: 10.3390/ijms21186463. Int J Mol Sci. 2020. PMID: 32899717 Free PMC article.
-
Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis.J Neuroinflammation. 2016 Aug 26;13(1):207. doi: 10.1186/s12974-016-0686-4. J Neuroinflammation. 2016. PMID: 27566665 Free PMC article.
-
Does Siponimod Exert Direct Effects in the Central Nervous System?Cells. 2020 Jul 24;9(8):1771. doi: 10.3390/cells9081771. Cells. 2020. PMID: 32722245 Free PMC article. Review.
-
The Benefits and Risks of Switching from Fingolimod to Siponimod for the Treatment of Relapsing-Remitting and Secondary Progressive Multiple Sclerosis.Drugs R D. 2023 Dec;23(4):331-338. doi: 10.1007/s40268-023-00434-6. Epub 2023 Aug 28. Drugs R D. 2023. PMID: 37640862 Free PMC article. Review.
References
-
- Walton C., King R., Rechtman L., Kaye W., Leray E., Marrie R.A., Robertson N., La Rocca N., Uitdehaag B., van der Mei I., et al. Rising Prevalence of Multiple Sclerosis Worldwide: Insights from the Atlas of MS, Third Edition. Mult. Scler. 2020;26:1816–1821. doi: 10.1177/1352458520970841. - DOI - PMC - PubMed
-
- Bjornevik K., Cortese M., Healy B.C., Kuhle J., Mina M.J., Leng Y., Elledge S.J., Niebuhr D.W., Scher A.I., Munger K.L., et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science. 2022;375:296–301. doi: 10.1126/science.abj8222. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
