

Variation in the ACE2 receptor has limited utility for SARS-CoV-2 host prediction
- PMID: 36416537
- PMCID: PMC9683784
- DOI: 10.7554/eLife.80329
Variation in the ACE2 receptor has limited utility for SARS-CoV-2 host prediction
Abstract
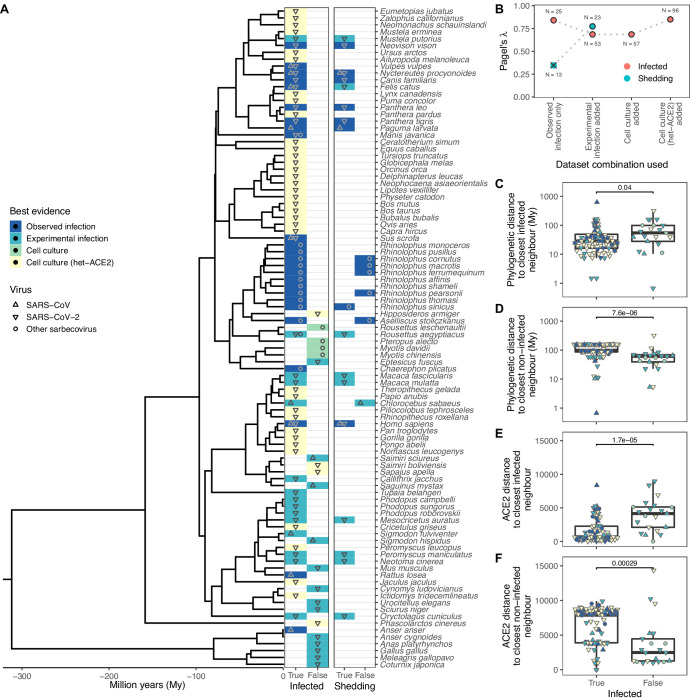
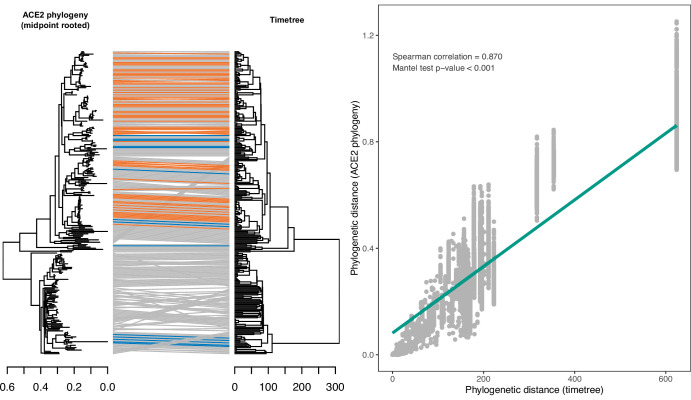
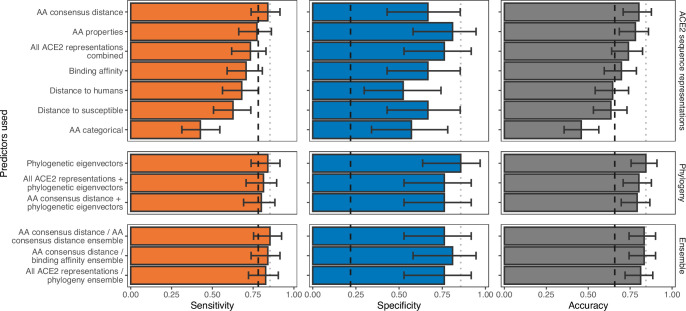
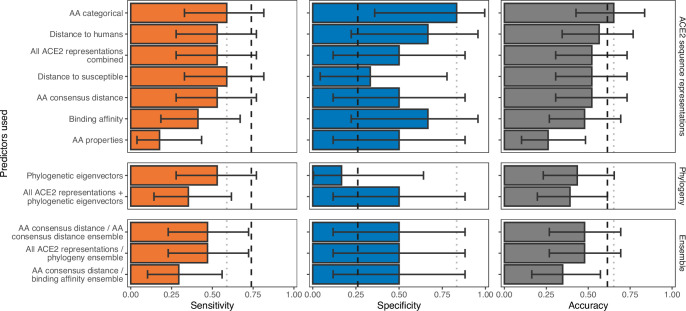
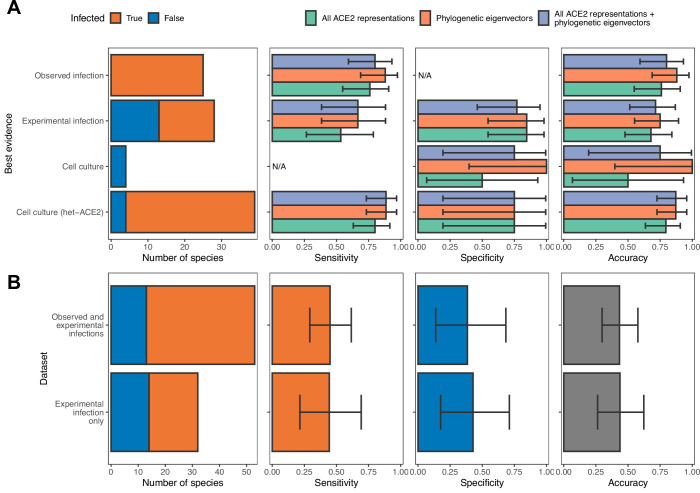
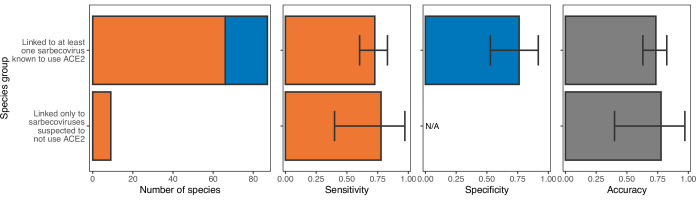
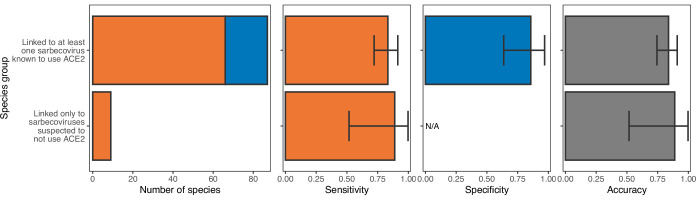
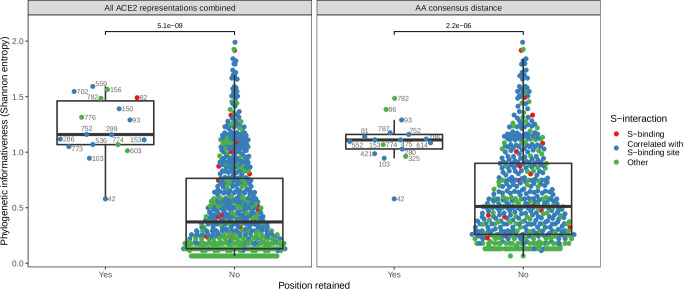
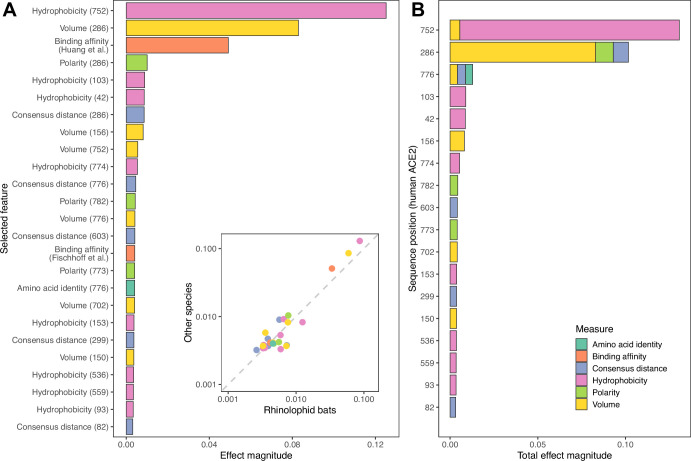
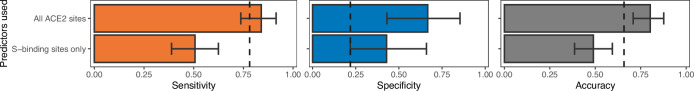
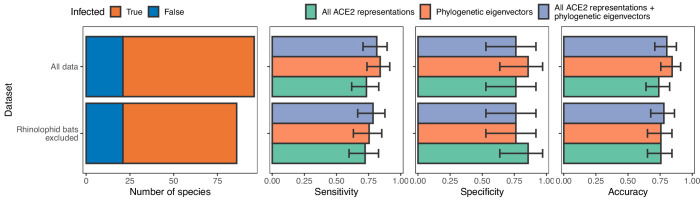
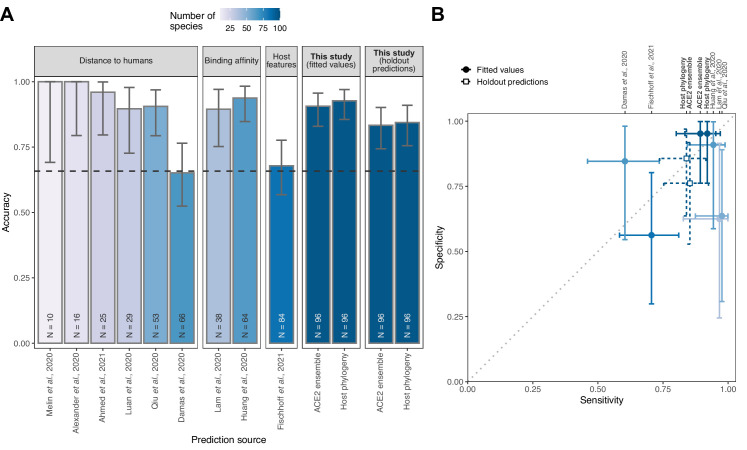
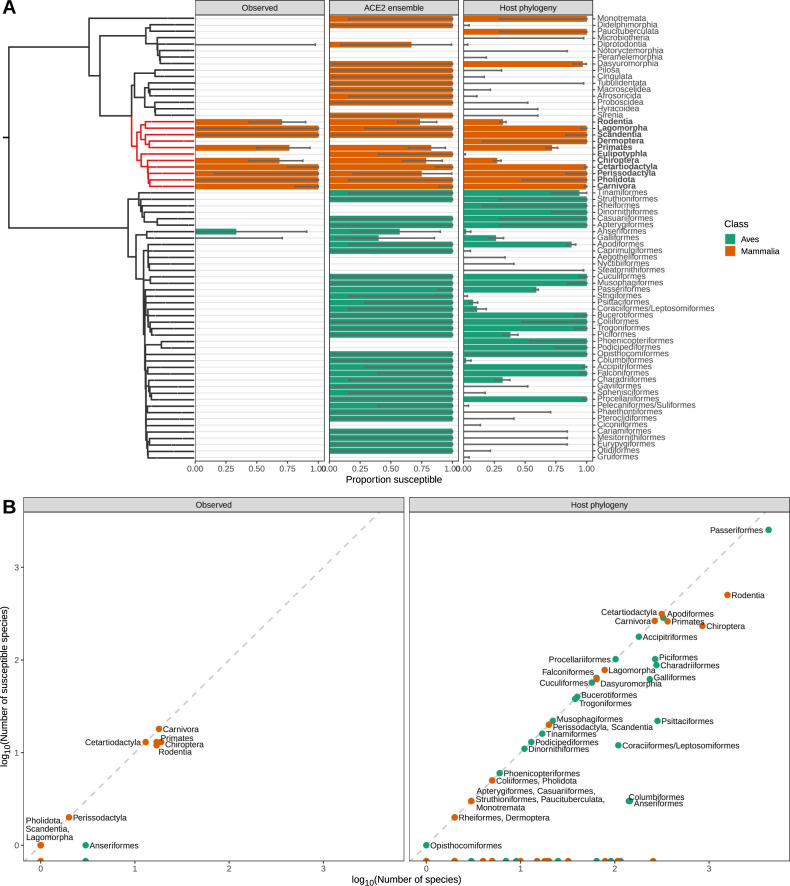
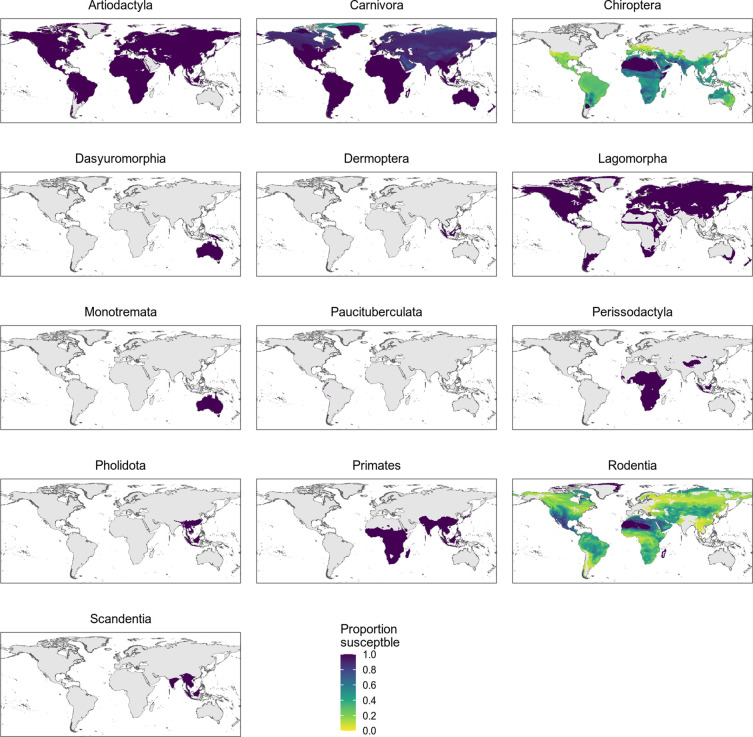
Transmission of SARS-CoV-2 from humans to other species threatens wildlife conservation and may create novel sources of viral diversity for future zoonotic transmission. A variety of computational heuristics have been developed to pre-emptively identify susceptible host species based on variation in the angiotensin-converting enzyme 2 (ACE2) receptor used for viral entry. However, the predictive performance of these heuristics remains unknown. Using a newly compiled database of 96 species, we show that, while variation in ACE2 can be used by machine learning models to accurately predict animal susceptibility to sarbecoviruses (accuracy = 80.2%, binomial confidence interval [CI]: 70.8-87.6%), the sites informing predictions have no known involvement in virus binding and instead recapitulate host phylogeny. Models trained on host phylogeny alone performed equally well (accuracy = 84.4%, CI: 75.5-91.0%) and at a level equivalent to retrospective assessments of accuracy for previously published models. These results suggest that the predictive power of ACE2-based models derives from strong correlations with host phylogeny rather than processes which can be mechanistically linked to infection biology. Further, biased availability of ACE2 sequences misleads projections of the number and geographic distribution of at-risk species. Models based on host phylogeny reduce this bias, but identify a very large number of susceptible species, implying that model predictions must be combined with local knowledge of exposure risk to practically guide surveillance. Identifying barriers to viral infection or onward transmission beyond receptor binding and incorporating data which are independent of host phylogeny will be necessary to manage the ongoing risk of establishment of novel animal reservoirs of SARS-CoV-2.
Keywords: ACE2; SARS-CoV-2; ecology; host range; infectious disease; microbiology; viruses.
Plain language summary
The COVID-19 pandemic affects humans, but also many of the animals we interact with. So far, humans have transmitted the SARS-CoV-2 virus to pet dogs and cats, a wide range of zoo animals, and even wildlife. Transmission of SARS-CoV-2 from humans to animals can lead to outbreaks amongst certain species, which can endanger animal populations and create new sources of human infections. Thus, careful monitoring of animal infections may help protect both animals and humans. Identifying which animals are susceptible to SARS-CoV-2 would help scientists monitor these species for outbreaks and viral circulation. Unfortunately, testing whether SARS-CoV-2 can infect different species in the laboratory is both time-consuming and expensive. To overcome this obstacle, researchers have used computational methods and existing data about the structure and genetic sequences of ACE2 receptors – the proteins on the cell surface that SARS-CoV-2 uses to enter the cell – to predict SARS-COV-2 susceptibility in different species. However, it remained unclear how accurate this approach was at predicting susceptibility in different animals, or whether their correct predictions indicated causal links between ACE2 variability and SARS-CoV-2 susceptibility. To assess the usefulness of this approach, Mollentze et al. started by using data on the ACE2 receptors from 96 different species and building a machine learning model to predict how susceptible those species might be to SARS-CoV-2. The susceptibility of these species had either been observed in natural infections – in zoos, for example – or had been assessed in the laboratory, so Mollentze et al. were able to use this information to determine how good both their model and previous approaches based on the sequence of ACE2 receptors were. The results showed that while the model was quite accurate (it correctly predicted susceptibility to SARS-CoV-2 about 80% of the time), its predictions were based on regions of the ACE2 receptors that were not known to interact with the virus. Instead, the regions that the machine learning model relied on were ones that tend to vary more the more distantly related two species are. This indicates that existing computational approaches are likely not relying on information about how ACE2 receptors interact with SARS-CoV-2 to predict susceptibility. Instead, they are simply using information on how closely related the different animal species are, which is much easier to source than data about ACE2 receptors. Indeed, the sequences of the ACE2 receptors in many species are unknown and the species for which this information is available come only from a few geographic areas. Mollentze et al. also showed that limiting the predictions about susceptibility to these species could mislead scientists when deciding which species and geographic areas to surveil for possible viral circulation. Instead, it may be more effective and cost-efficient to use animal relatedness to predict susceptibility to SARS-CoV-2. This makes it possible to make predictions for nearly all mammals, while being just as accurate as models based on ACE2 receptor data. However, Mollentze et al. point out that this approach would still fail to narrow down the number of animals that need to be monitored enough for it to be practical. Considering additional factors like how often the animals interact with humans or how prone they are to transmit the virus among themselves may help narrow it down more. Further research is therefore needed to identify the best multifactor approaches to identifying which animal populations should be monitored.
© 2022, Mollentze et al.
Conflict of interest statement
NM, DK, UM, RB, DS No competing interests declared
Figures




Similar articles
-
Functional and genetic analysis of viral receptor ACE2 orthologs reveals a broad potential host range of SARS-CoV-2.Proc Natl Acad Sci U S A. 2021 Mar 23;118(12):e2025373118. doi: 10.1073/pnas.2025373118. Proc Natl Acad Sci U S A. 2021. PMID: 33658332 Free PMC article.
-
Predicting susceptibility to SARS-CoV-2 infection based on structural differences in ACE2 across species.FASEB J. 2020 Dec;34(12):15946-15960. doi: 10.1096/fj.202001808R. Epub 2020 Oct 4. FASEB J. 2020. PMID: 33015868 Free PMC article.
-
Variants in ACE2; potential influences on virus infection and COVID-19 severity.Infect Genet Evol. 2021 Jun;90:104773. doi: 10.1016/j.meegid.2021.104773. Epub 2021 Feb 17. Infect Genet Evol. 2021. PMID: 33607284 Free PMC article. Review.
-
Broad and Differential Animal Angiotensin-Converting Enzyme 2 Receptor Usage by SARS-CoV-2.J Virol. 2020 Aug 31;94(18):e00940-20. doi: 10.1128/JVI.00940-20. Print 2020 Aug 31. J Virol. 2020. PMID: 32661139 Free PMC article.
-
Contributions of human ACE2 and TMPRSS2 in determining host-pathogen interaction of COVID-19.J Genet. 2021;100(1):12. doi: 10.1007/s12041-021-01262-w. J Genet. 2021. PMID: 33707363 Free PMC article. Review.
Cited by
-
An Outbreak of SARS-CoV-2 in Captive Armadillos Associated with Gamma Variant in Argentina.Ecohealth. 2024 Dec;21(2-4):183-194. doi: 10.1007/s10393-024-01686-7. Epub 2024 Jun 6. Ecohealth. 2024. PMID: 38844740
-
Heterogeneities in infection outcomes across species: sex and tissue differences in virus susceptibility.Peer Community J. 2023 Feb 7;3:pcjournal.242. doi: 10.24072/pcjournal.242. Peer Community J. 2023. PMID: 36811030 Free PMC article.
-
SARS-CoV-2 Surveillance between 2020 and 2021 of All Mammalian Species in Two Flemish Zoos (Antwerp Zoo and Planckendael Zoo).Vet Sci. 2023 May 31;10(6):382. doi: 10.3390/vetsci10060382. Vet Sci. 2023. PMID: 37368768 Free PMC article.
-
Distinct phenotype of SARS-CoV-2 Omicron BA.1 in human primary cells but no increased host range in cell lines of putative mammalian reservoir species.Virus Res. 2024 Jan 2;339:199255. doi: 10.1016/j.virusres.2023.199255. Epub 2023 Nov 6. Virus Res. 2024. PMID: 38389324 Free PMC article.
References
-
- Allender MC, Adkesson MJ, Langan JN, Delk KW, Meehan T, Aitken-Palmer C, McEntire MM, Killian ML, Torchetti M, Morales SA, Austin C, Fredrickson R, Olmstead C, Ke R, Smith R, Hostnik ET, Terio K, Wang L. Multi-species outbreak of SARS-CoV-2 delta variant in a zoological institution, with the detection in two new families of carnivores. Transboundary and Emerging Diseases. 2022;69:e3060–e3075. doi: 10.1111/tbed.14662. - DOI - PMC - PubMed
-
- Baker FB. Stability of two hierarchical grouping techniques case 1: sensitivity to data errors. Journal of the American Statistical Association. 1974;69:440. doi: 10.2307/2285675. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
