

Oncolytic virus driven T-cell-based combination immunotherapy platform for colorectal cancer
- PMID: 36405739
- PMCID: PMC9670134
- DOI: 10.3389/fimmu.2022.1029269
Oncolytic virus driven T-cell-based combination immunotherapy platform for colorectal cancer
Abstract
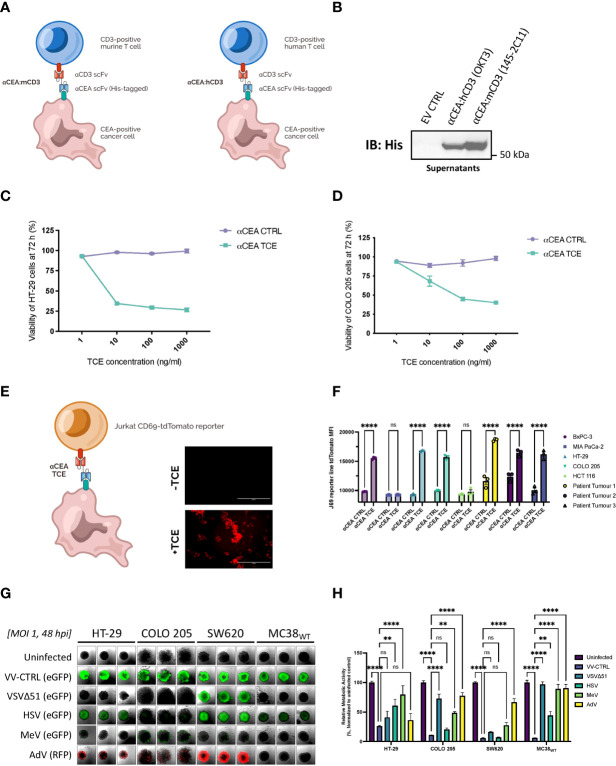
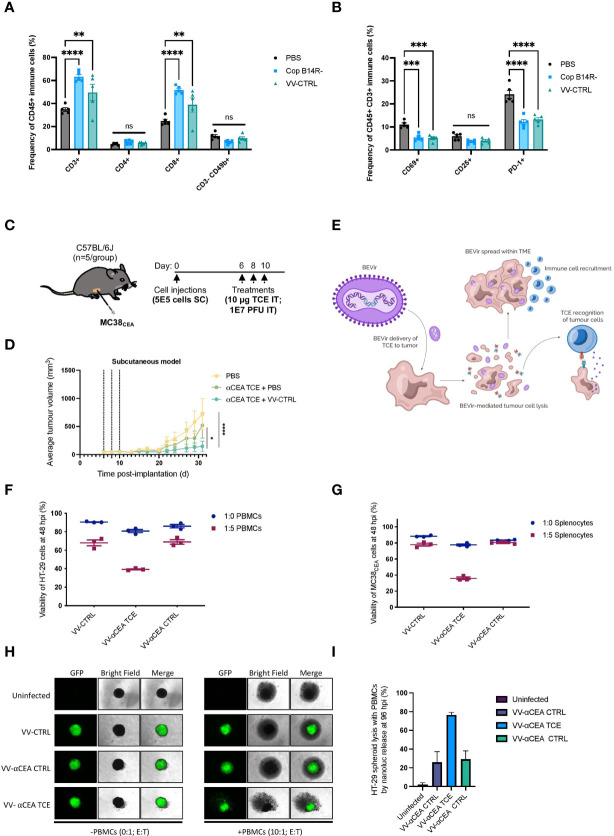
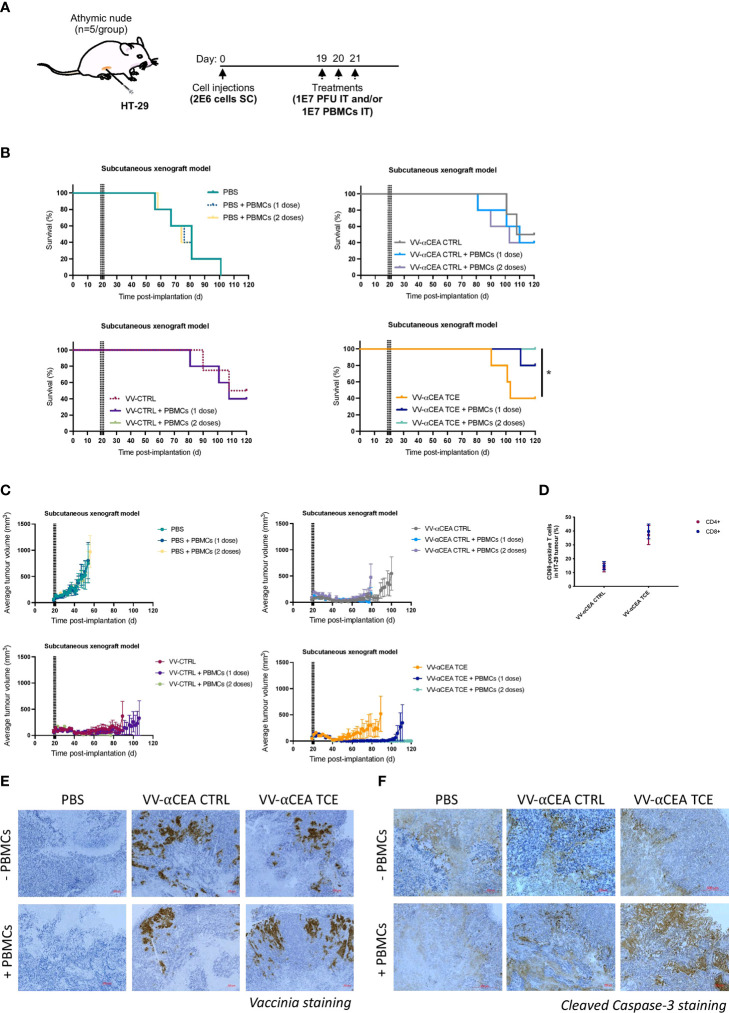
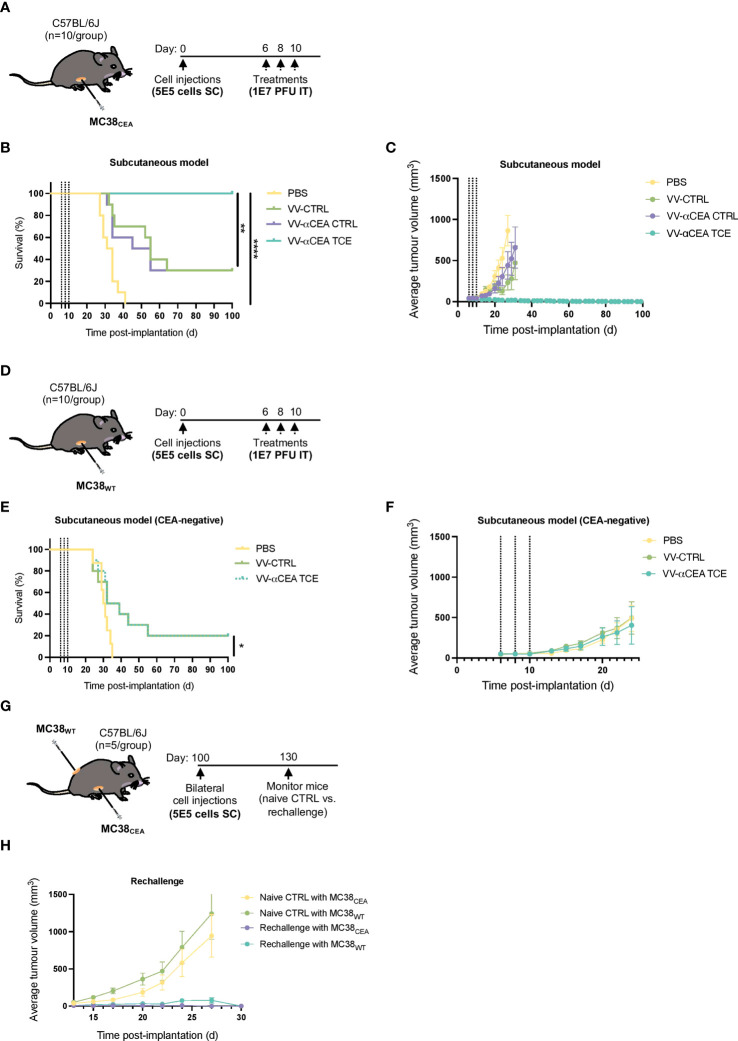
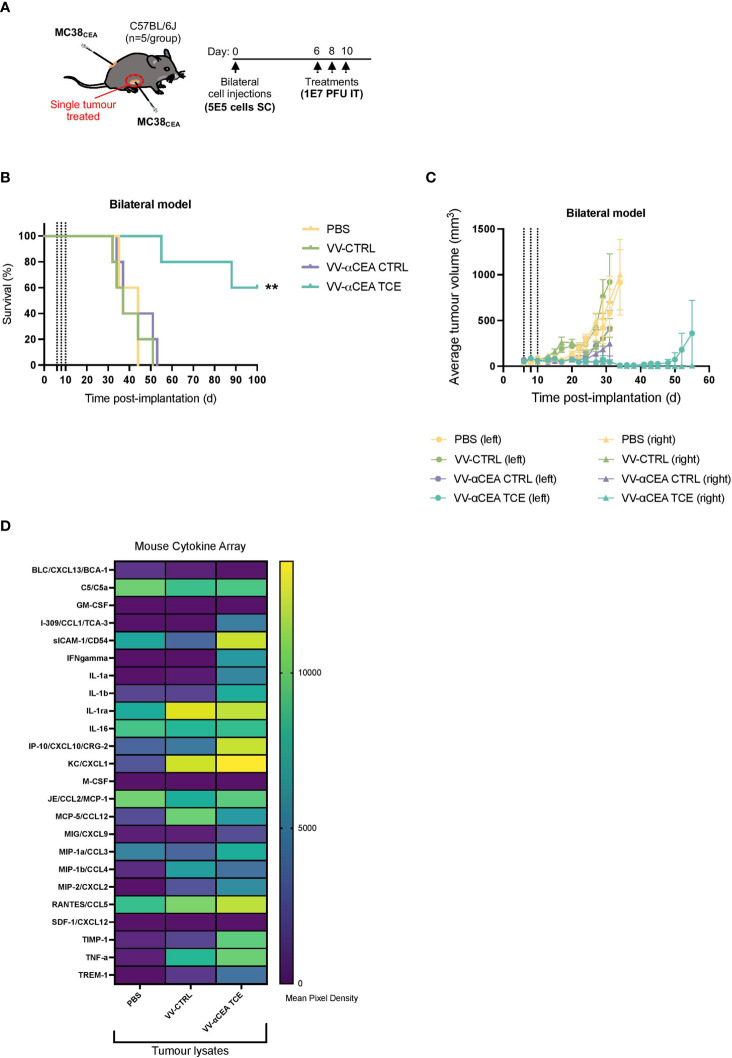
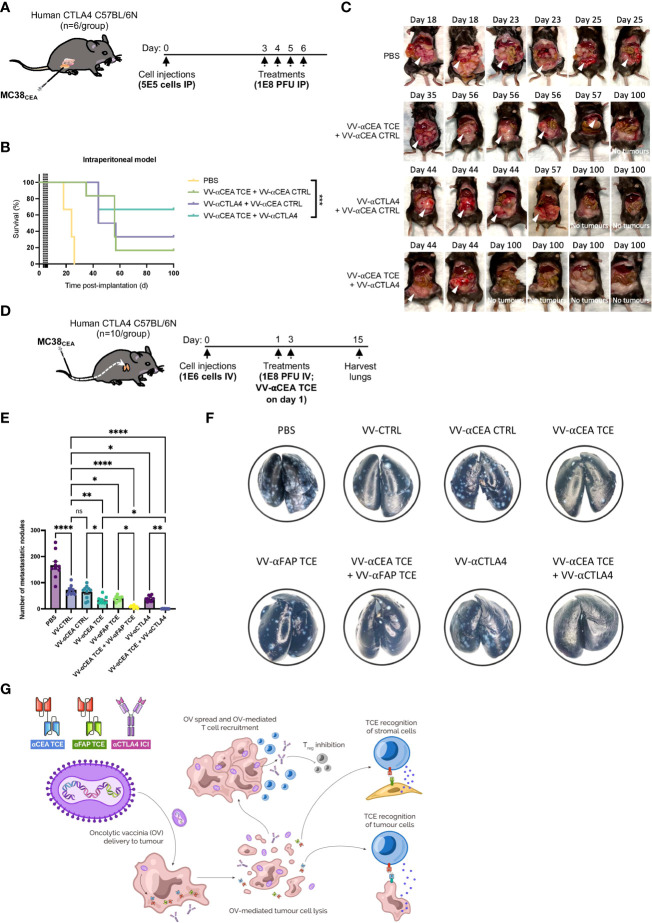
Colorectal cancer is the third most diagnosed cancer and the second leading cause of cancer mortality worldwide, highlighting an urgent need for new therapeutic options and combination strategies for patients. The orchestration of potent T cell responses against human cancers is necessary for effective antitumour immunity. However, regression of a limited number of cancers has been induced by immune checkpoint inhibitors, T cell engagers (TCEs) and/or oncolytic viruses. Although one TCE has been FDA-approved for the treatment of hematological malignancies, many challenges exist for the treatment of solid cancers. Here, we show that TCEs targeting CEACAM5 and CD3 stimulate robust activation of CD4 and CD8-positive T cells in in vitro co-culture models with colorectal cancer cells, but in vivo efficacy is hindered by a lack of TCE retention in the tumour microenvironment and short TCE half-life, as demonstrated by HiBiT bioluminescent TCE-tagging technology. To overcome these limitations, we engineered Bispecific Engager Viruses, or BEVirs, a novel tumour-targeted vaccinia virus platform for intra-tumour delivery of these immunomodulatory molecules. We characterized virus-mediated TCE-secretion, TCE specificity and functionality from infected colorectal cancer cells and patient tumour samples, as well as TCE cytotoxicity in spheroid models, in the presence and absence of T cells. Importantly, we show regression of colorectal tumours in both syngeneic and xenograft mouse models. Our data suggest that a different profile of cytokines may contribute to the pro-inflammatory and immune effects driven by T cells in the tumour microenvironment to provide long-lasting immunity and abscopal effects. We establish combination regimens with immune checkpoint inhibitors for aggressive colorectal peritoneal metastases. We also observe a significant reduction in lung metastases of colorectal tumours through intravenous delivery of our oncolytic virus driven T-cell based combination immunotherapy to target colorectal tumours and FAP-positive stromal cells or CTLA4-positive Treg cells in the tumour microenvironment. In summary, we devised a novel combination strategy for the treatment of colorectal cancers using oncolytic vaccinia virus to enhance immune-payload delivery and boost T cell responses within tumours.
Keywords: CEA; CTLA4; FAP; T cell engager; oncolytic virus.
Copyright © 2022 Crupi, Taha, Janssen, Petryk, Boulton, Alluqmani, Jirovec, Kassas, Khan, Vallati, Lee, Huang, Huh, Pikor, He, Marius, Austin, Duong, Pelin, Neault, Azad, Breitbach, Stojdl, Burgess, McComb, Auer, Diallo, Ilkow and Bell.
Conflict of interest statement
We declare that JB has an interest in Turnstone Biologics, which develops the oncolytic vaccinia virus as an OV platform. LP, MH, JD, AP, CB, DS and MB have worked for Turnstone Biologics. JB, CB, DS and MB are shareholders in Turnstone Biologics. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
An engineered oncolytic vaccinia virus encoding a single-chain variable fragment against TIGIT induces effective antitumor immunity and synergizes with PD-1 or LAG-3 blockade.J Immunother Cancer. 2021 Dec;9(12):e002843. doi: 10.1136/jitc-2021-002843. J Immunother Cancer. 2021. PMID: 34949694 Free PMC article.
-
Enhanced antitumor efficacy of a novel oncolytic vaccinia virus encoding a fully monoclonal antibody against T-cell immunoglobulin and ITIM domain (TIGIT).EBioMedicine. 2021 Feb;64:103240. doi: 10.1016/j.ebiom.2021.103240. Epub 2021 Feb 10. EBioMedicine. 2021. PMID: 33581644 Free PMC article.
-
Oncolytic virus expressing PD-1 inhibitors activates a collaborative intratumoral immune response to control tumor and synergizes with CTLA-4 or TIM-3 blockade.J Immunother Cancer. 2022 Jun;10(6):e004762. doi: 10.1136/jitc-2022-004762. J Immunother Cancer. 2022. PMID: 35688558 Free PMC article.
-
New hopes for the breast cancer treatment: perspectives on the oncolytic virus therapy.Front Immunol. 2024 Mar 21;15:1375433. doi: 10.3389/fimmu.2024.1375433. eCollection 2024. Front Immunol. 2024. PMID: 38576614 Free PMC article. Review.
-
Oncolytic viruses encoding bispecific T cell engagers: a blueprint for emerging immunovirotherapies.J Hematol Oncol. 2021 Apr 16;14(1):63. doi: 10.1186/s13045-021-01075-5. J Hematol Oncol. 2021. PMID: 33863363 Free PMC article. Review.
Cited by
-
Tumor microenvironment-modulating oncolytic adenovirus combined with GSK-3β inhibitor enhances antitumor immune response against bladder cancer.Front Immunol. 2024 May 15;15:1360436. doi: 10.3389/fimmu.2024.1360436. eCollection 2024. Front Immunol. 2024. PMID: 38812516 Free PMC article.
-
Perioperative immune checkpoint inhibition for colorectal cancer: recent advances and future directions.Front Immunol. 2023 Nov 13;14:1269341. doi: 10.3389/fimmu.2023.1269341. eCollection 2023. Front Immunol. 2023. PMID: 38022667 Free PMC article. Review.
-
Extracellular Vesicles and Viruses: Two Intertwined Entities.Int J Mol Sci. 2023 Jan 5;24(2):1036. doi: 10.3390/ijms24021036. Int J Mol Sci. 2023. PMID: 36674550 Free PMC article. Review.
-
Construction and application of adenoviral vectors.Mol Ther Nucleic Acids. 2023 Sep 9;34:102027. doi: 10.1016/j.omtn.2023.09.004. eCollection 2023 Dec 12. Mol Ther Nucleic Acids. 2023. PMID: 37808925 Free PMC article. Review.
-
Immune landscape and response to oncolytic virus-based immunotherapy.Front Med. 2024 Jun;18(3):411-429. doi: 10.1007/s11684-023-1048-0. Epub 2024 Mar 8. Front Med. 2024. PMID: 38453818 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous