

Molecular Genetics of GLUT1DS Italian Pediatric Cohort: 10 Novel Disease-Related Variants and Structural Analysis
- PMID: 36362347
- PMCID: PMC9654628
- DOI: 10.3390/ijms232113560
Molecular Genetics of GLUT1DS Italian Pediatric Cohort: 10 Novel Disease-Related Variants and Structural Analysis
Abstract
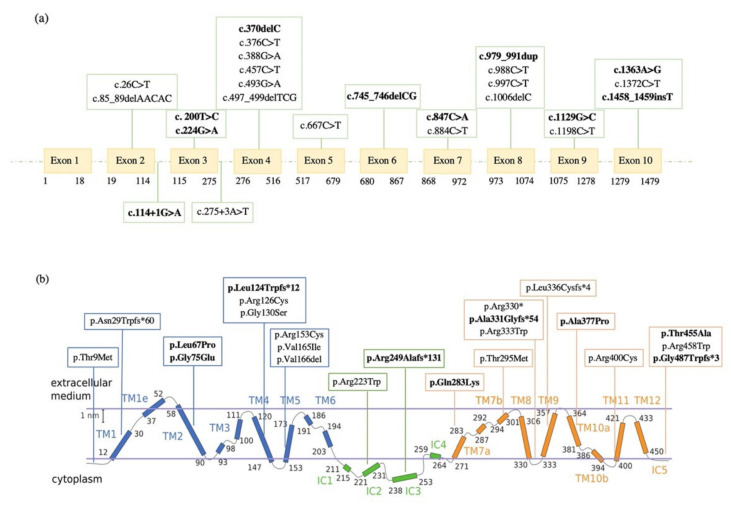
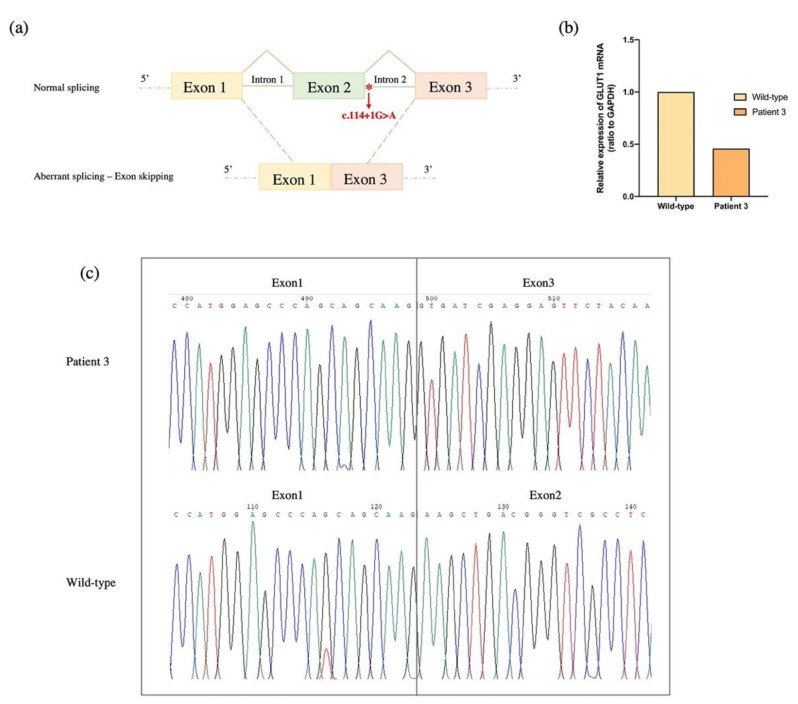
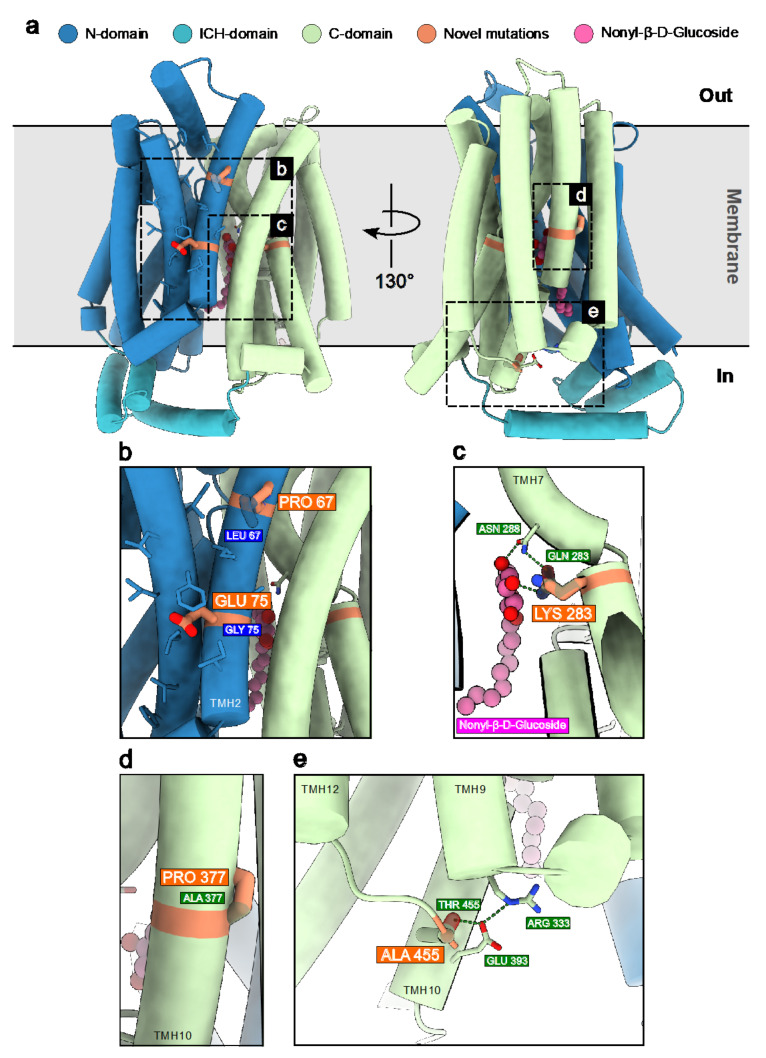
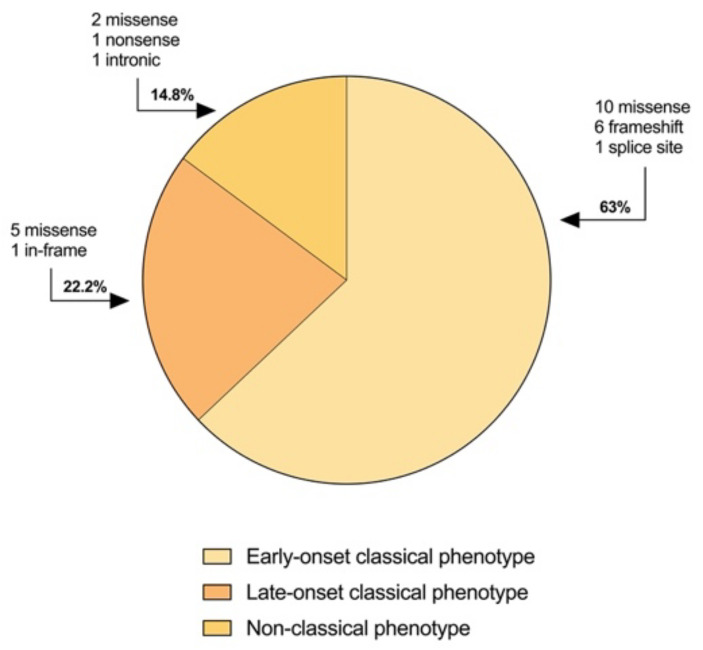
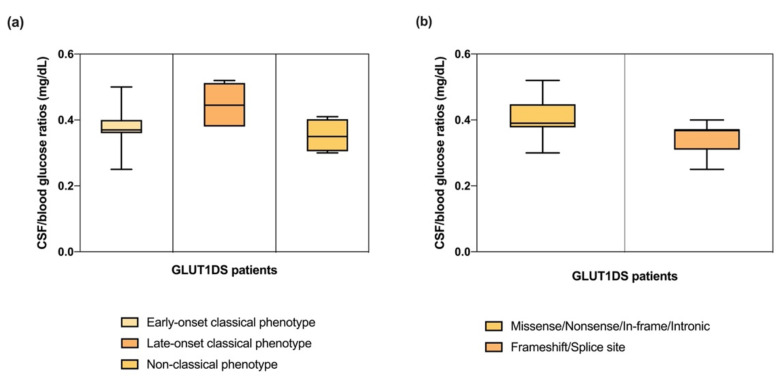
GLUT1 deficiency syndrome (GLUT1DS1; OMIM #606777) is a rare genetic metabolic disease, characterized by infantile-onset epileptic encephalopathy, global developmental delay, progressive microcephaly, and movement disorders (e.g., spasticity and dystonia). It is caused by heterozygous mutations in the SLC2A1 gene, which encodes the GLUT1 protein, a glucose transporter across the blood-brain barrier (BBB). Most commonly, these variants arise de novo resulting in sporadic cases, although several familial cases with AD inheritance pattern have been described. Twenty-seven Italian pediatric patients, clinically suspect of GLUT1DS from both sporadic and familial cases, have been enrolled. We detected by trios sequencing analysis 25 different variants causing GLUT1DS. Of these, 40% of the identified variants (10 out of 25) had never been reported before, including missense, frameshift, and splice site variants. Their structural mapping on the X-ray structure of GLUT1 strongly suggested the potential pathogenic effects of these novel disease-related mutations, broadening the genotypic spectrum heterogeneity found in the SLC2A1 gene. Moreover, 24% is located in a vulnerable region of the GLUT1 protein that involves transmembrane 4 and 5 helices encoded by exon 4, confirming a mutational hotspot in the SLC2A1 gene. Lastly, we investigated possible correlations between mutation type and clinical and biochemical data observed in our GLUT1DS cohort, revealing that splice site and frameshift variants are related to a more severe phenotype and low CSF parameters.
Keywords: GLUT1 deficiency syndrome; GLUT1 structure analysis; novel SLC2A1 variants.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Sporadic and familial glut1ds Italian patients: A wide clinical variability.Seizure. 2015 Jan;24:28-32. doi: 10.1016/j.seizure.2014.11.009. Epub 2014 Nov 26. Seizure. 2015. PMID: 25564316
-
The clinical and biochemical hallmarks generally associated with GLUT1DS may be caused by defects in genes other than SLC2A1.Clin Genet. 2022 Jul;102(1):40-55. doi: 10.1111/cge.14138. Epub 2022 Apr 15. Clin Genet. 2022. PMID: 35388452 Free PMC article.
-
Re-analysis of whole-exome sequencing data reveals a novel splicing variant in the SLC2A1 in a patient with GLUT1 Deficiency Syndrome 1 accompanied by hemangioma: a case report.BMC Med Genomics. 2021 Jul 31;14(1):197. doi: 10.1186/s12920-021-01045-3. BMC Med Genomics. 2021. PMID: 34332575 Free PMC article.
-
GLUT1 Deficiency Syndrome-Early Treatment Maintains Cognitive Development? (Literature Review and Case Report).Genes (Basel). 2021 Aug 31;12(9):1379. doi: 10.3390/genes12091379. Genes (Basel). 2021. PMID: 34573360 Free PMC article. Review.
-
GLUT1 deficiency syndrome 2013: current state of the art.Seizure. 2013 Dec;22(10):803-11. doi: 10.1016/j.seizure.2013.07.003. Epub 2013 Jul 26. Seizure. 2013. PMID: 23890838 Review.
Cited by
-
Clinical and genetic analysis of children with glucose transporter type 1 deficiency syndrome.Med Int (Lond). 2024 Jul 19;4(6):57. doi: 10.3892/mi.2024.181. eCollection 2024 Nov-Dec. Med Int (Lond). 2024. PMID: 39092009 Free PMC article.
-
Expression assay of calcium signaling related lncRNAs in autism.Mol Biol Rep. 2024 Jan 24;51(1):185. doi: 10.1007/s11033-023-09182-x. Mol Biol Rep. 2024. PMID: 38265729
-
The diagnostic and prognostic role of cerebrospinal fluid biomarkers in glucose transporter 1 deficiency: a systematic review.Eur J Pediatr. 2024 Sep;183(9):3665-3678. doi: 10.1007/s00431-024-05657-6. Epub 2024 Jul 2. Eur J Pediatr. 2024. PMID: 38954008 Free PMC article. Review.
References
-
- De Vivo D.C., Trifiletti R.R., Jacobson R.I., Ronen G.M., Behmand R.A., Harik S.I. Defective Glucose Transport across the Blood-Brain Barrier as a Cause of Persistent Hypoglycorrhachia, Seizures, and Developmental Delay. N. Engl. J. Med. 1991;325:703–709. doi: 10.1056/NEJM199109053251006. - DOI - PubMed
-
- De Vivo D.C., Leary L., Wang D. Glucose transporter 1 deficiency syndrome and other glycolytic defects. J. Child Neurol. 2002;17((Suppl. S3)):3S15-23. discussion 3S24-5. - PubMed
-
- Suls A., Dedeken P., Goffin K., Van Esch H., Dupont P., Cassiman D., Kempfle J., Wuttke T.V., Weber Y., Lerche H., et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. 2008;131:1831–1844. doi: 10.1093/brain/awn113. - DOI - PMC - PubMed
-
- Leen W.G., Klepper J., Verbeek M.M., Leferink M., Hofste T., Van Engelen B.G., Wevers R.A., Arthur T., Bahi-Buisson N., Ballhausen D., et al. Glucose transporter-1 deficiency syndrome: The expanding clinical and genetic spectrum of a treatable disorder. Brain. 2010;133:655–670. doi: 10.1093/brain/awp336. - DOI - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
