

MIR retrotransposons link the epigenome and the transcriptome of coding genes in acute myeloid leukemia
- PMID: 36316347
- PMCID: PMC9622910
- DOI: 10.1038/s41467-022-34211-x
MIR retrotransposons link the epigenome and the transcriptome of coding genes in acute myeloid leukemia
Abstract
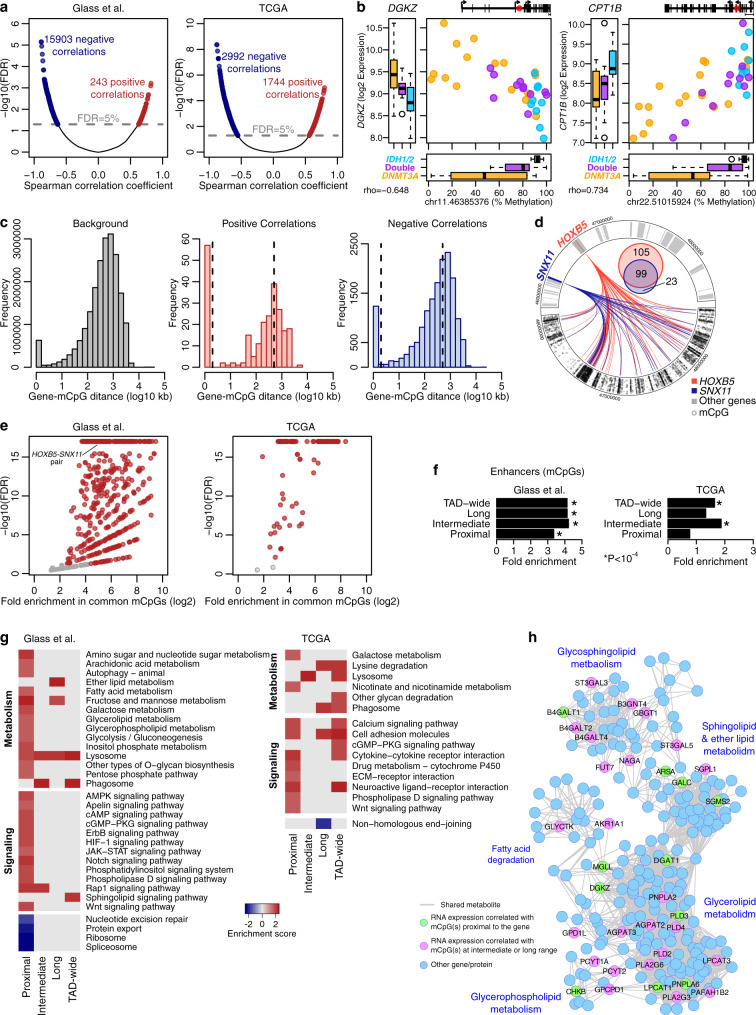
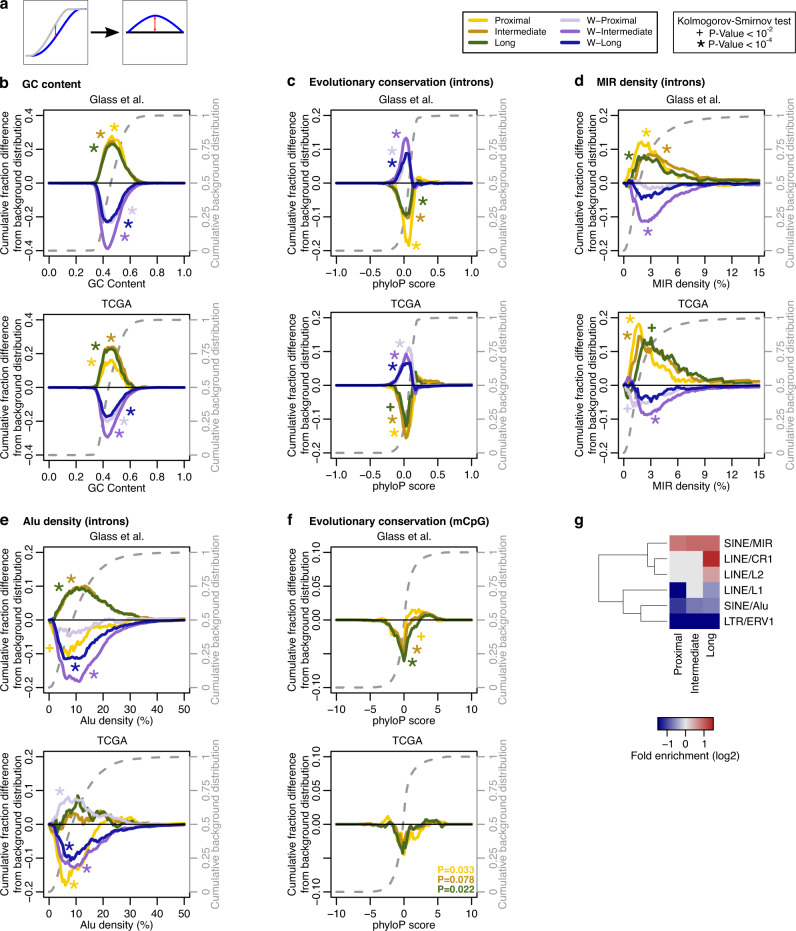
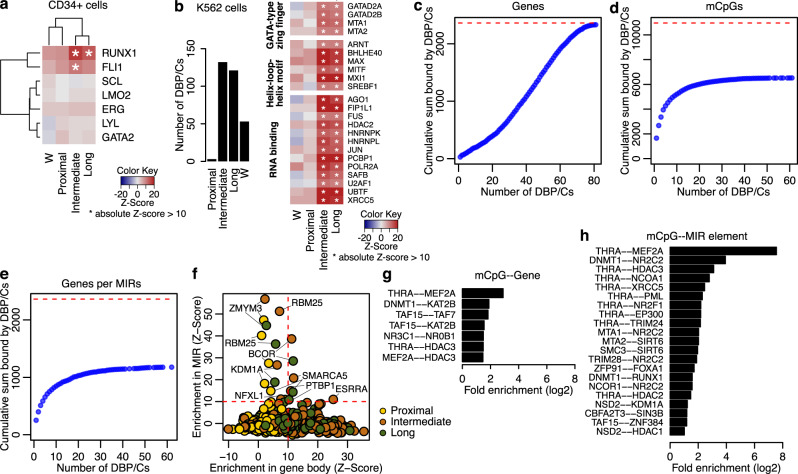
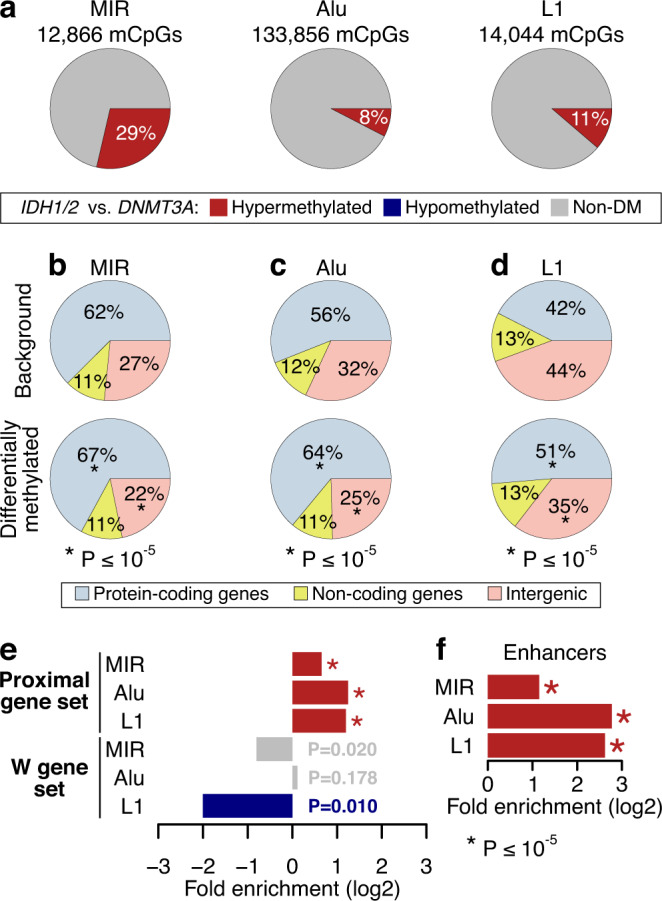
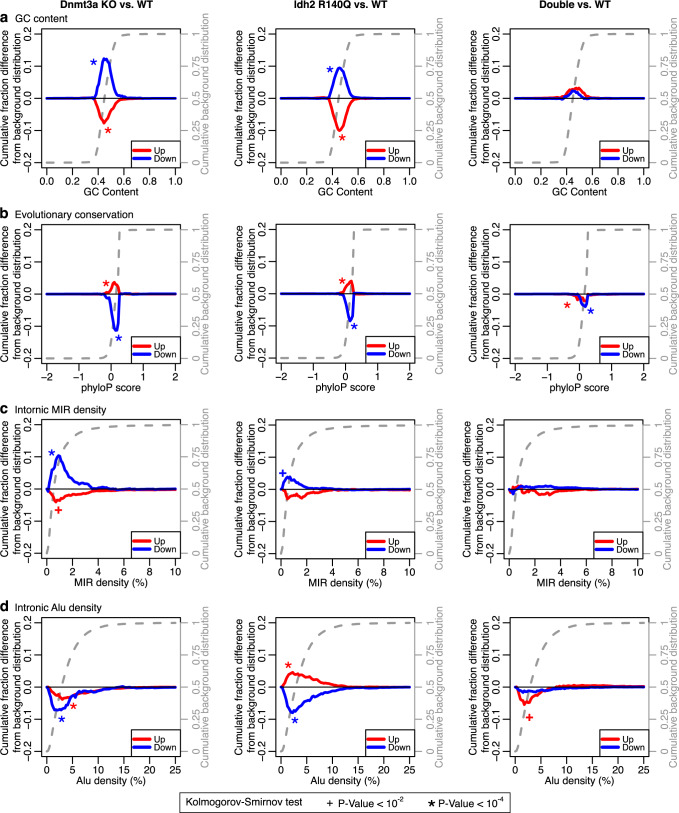
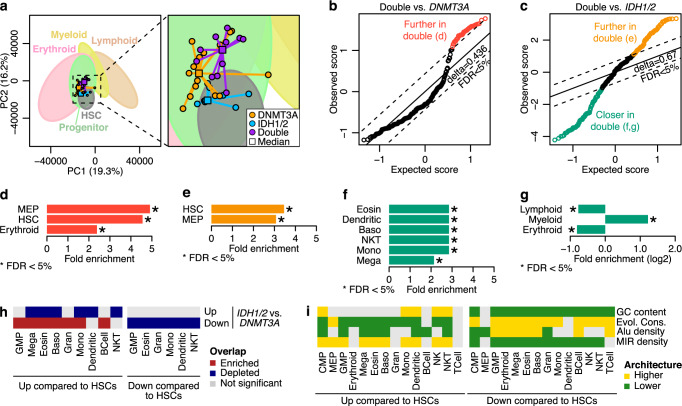
DNMT3A and IDH1/2 mutations combinatorically regulate the transcriptome and the epigenome in acute myeloid leukemia; yet the mechanisms of this interplay are unknown. Using a systems approach within topologically associating domains, we find that genes with significant expression-methylation correlations are enriched in signaling and metabolic pathways. The common denominator across these methylation-regulated genes is the density in MIR retrotransposons of their introns. Moreover, a discrete number of CpGs overlapping enhancers are responsible for regulating most of these genes. Established mouse models recapitulate the dependency of MIR-rich genes on the balanced expression of epigenetic modifiers, while projection of leukemic profiles onto normal hematopoiesis ones further consolidates the dependencies of methylation-regulated genes on MIRs. Collectively, MIR elements on genes and enhancers are susceptible to changes in DNA methylation activity and explain the cooperativity of proteins in this pathway in normal and malignant hematopoiesis.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Epigenetic Identity in AML Depends on Disruption of Nonpromoter Regulatory Elements and Is Affected by Antagonistic Effects of Mutations in Epigenetic Modifiers.Cancer Discov. 2017 Aug;7(8):868-883. doi: 10.1158/2159-8290.CD-16-1032. Epub 2017 Apr 13. Cancer Discov. 2017. PMID: 28408400 Free PMC article.
-
DNA methylation and hydroxymethylation patterns in acute myeloid leukemia patients with mutations in DNMT3A and IDH1/2 and their combinations.Cancer Biomark. 2019;25(1):43-51. doi: 10.3233/CBM-182176. Cancer Biomark. 2019. PMID: 30988238
-
Mutations in TET2 and DNMT3A genes are associated with changes in global and gene-specific methylation in acute myeloid leukemia.Tumour Biol. 2017 Oct;39(10):1010428317732181. doi: 10.1177/1010428317732181. Tumour Biol. 2017. PMID: 28992762
-
Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia.Int J Hematol. 2013 Dec;98(6):648-57. doi: 10.1007/s12185-013-1407-8. Epub 2013 Aug 15. Int J Hematol. 2013. PMID: 23949914 Free PMC article. Review.
-
[Functional role of DNMT3A mutation in acute myeloid leukemia].Rinsho Ketsueki. 2018;59(5):602-610. doi: 10.11406/rinketsu.59.602. Rinsho Ketsueki. 2018. PMID: 29877252 Review. Japanese.
Cited by
-
Research Progress on the Role of Epigenetic Methylation Modification in Hepatocellular Carcinoma.J Hepatocell Carcinoma. 2024 Jun 17;11:1143-1156. doi: 10.2147/JHC.S458734. eCollection 2024. J Hepatocell Carcinoma. 2024. PMID: 38911291 Free PMC article. Review.
References
-
- Glass JL, et al. Epigenetic identity in AML depends on disruption of nonpromoter regulatory elements and is affected by antagonistic effects of mutations in epigenetic modifiers. Cancer Discov. 2017;7:868–883. doi: 10.1158/2159-8290.CD-16-1032. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
