

Therapeutic Targeting of NF-κB in Acute Lung Injury: A Double-Edged Sword
- PMID: 36291185
- PMCID: PMC9601210
- DOI: 10.3390/cells11203317
Therapeutic Targeting of NF-κB in Acute Lung Injury: A Double-Edged Sword
Abstract
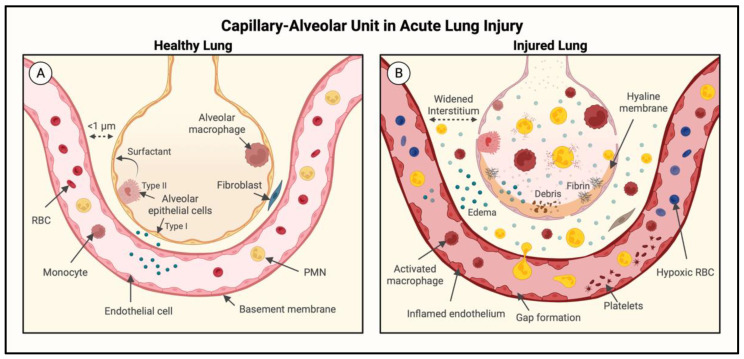
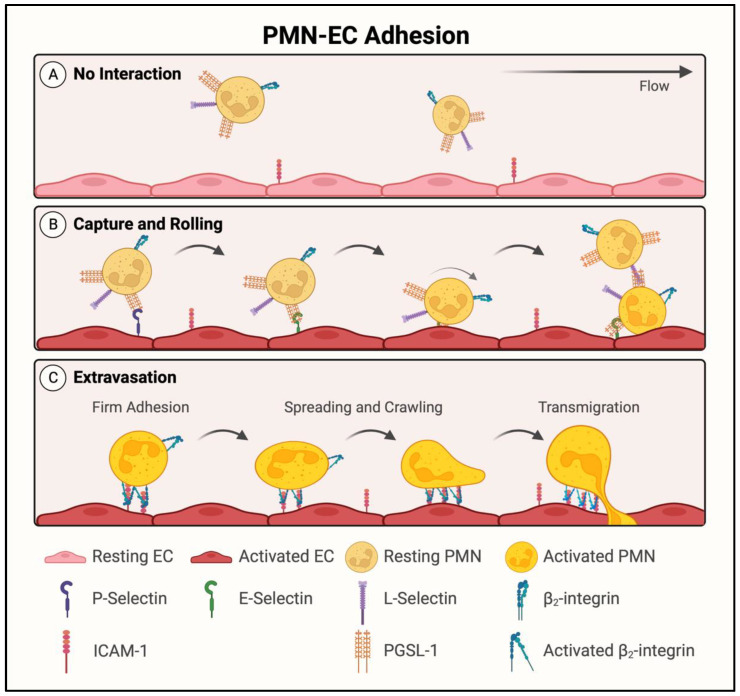
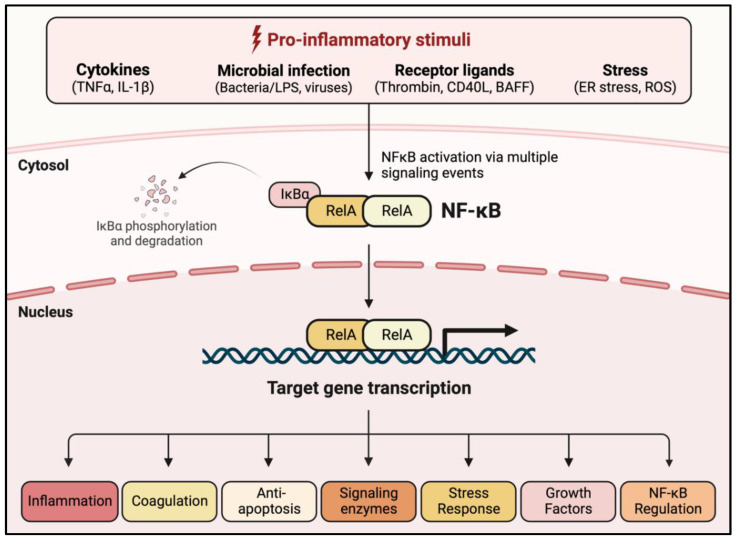
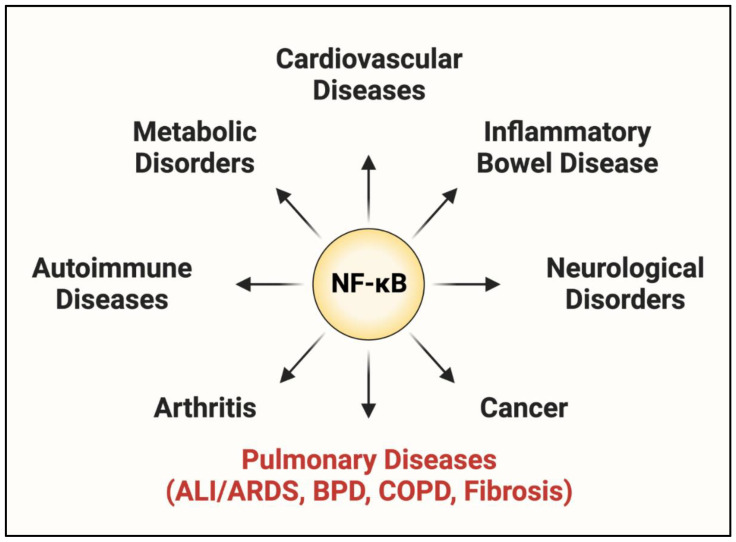
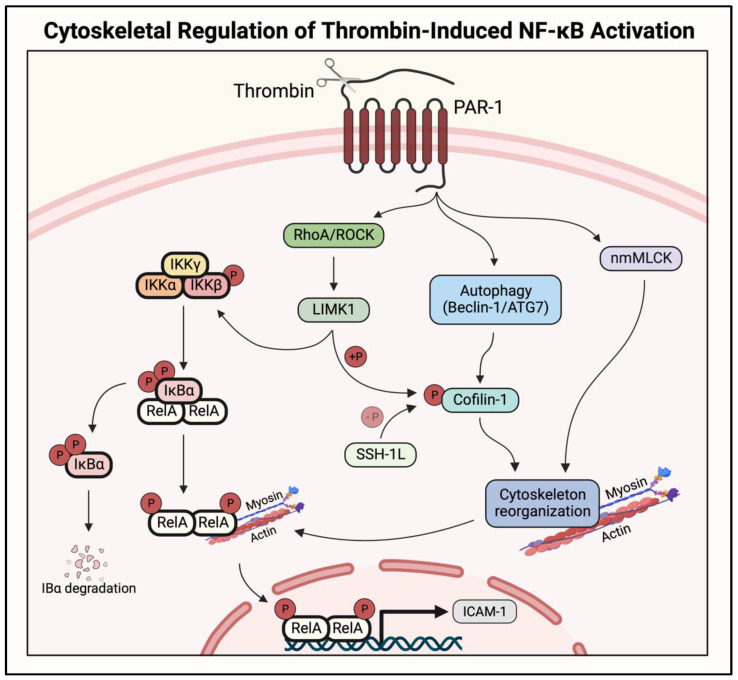
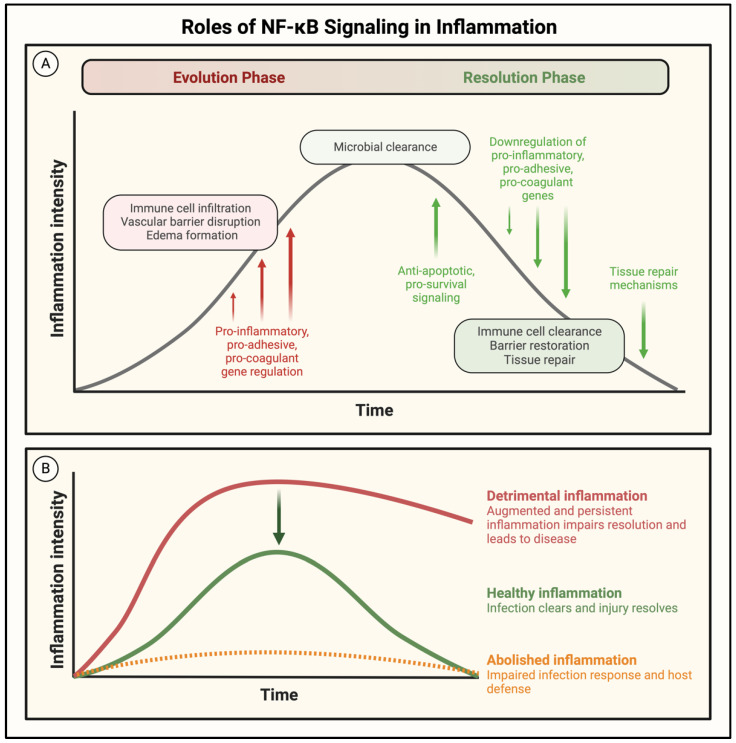
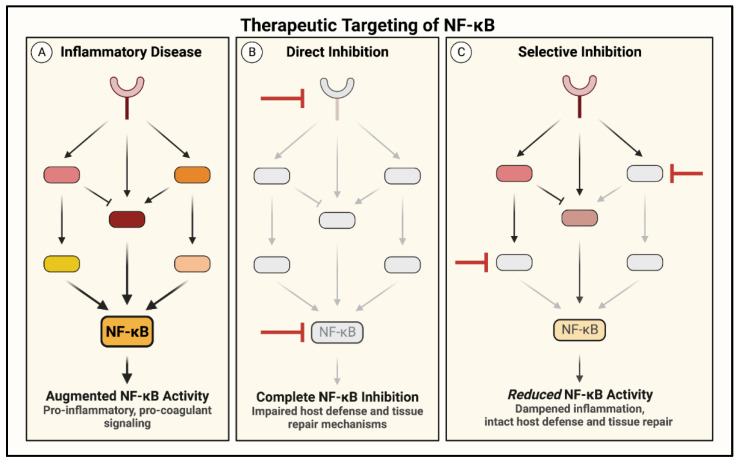
Acute lung injury/acute respiratory distress syndrome (ALI/ARDS) is a devastating disease that can be caused by a variety of conditions including pneumonia, sepsis, trauma, and most recently, COVID-19. Although our understanding of the mechanisms of ALI/ARDS pathogenesis and resolution has considerably increased in recent years, the mortality rate remains unacceptably high (~40%), primarily due to the lack of effective therapies for ALI/ARDS. Dysregulated inflammation, as characterized by massive infiltration of polymorphonuclear leukocytes (PMNs) into the airspace and the associated damage of the capillary-alveolar barrier leading to pulmonary edema and hypoxemia, is a major hallmark of ALI/ARDS. Endothelial cells (ECs), the inner lining of blood vessels, are important cellular orchestrators of PMN infiltration in the lung. Nuclear factor-kappa B (NF-κB) plays an essential role in rendering the endothelium permissive for PMN adhesion and transmigration to reach the inflammatory site. Thus, targeting NF-κB in the endothelium provides an attractive approach to mitigate PMN-mediated vascular injury, not only in ALI/ARDS, but in other inflammatory diseases as well in which EC dysfunction is a major pathogenic mechanism. This review discusses the role and regulation of NF-κB in the context of EC inflammation and evaluates the potential and problems of targeting it as a therapy for ALI/ARDS.
Keywords: endothelial cells; lung inflammatory injury; signal transduction; transcription factors.
Conflict of interest statement
The authors do not claim any conflicts of interest.
Figures
Similar articles
-
Protective effect of olopatadine hydrochloride against LPS-induced acute lung injury: via targeting NF-κB signaling pathway.Inflammopharmacology. 2024 Feb;32(1):603-627. doi: 10.1007/s10787-023-01353-3. Epub 2023 Oct 17. Inflammopharmacology. 2024. PMID: 37847473
-
The HDL from septic-ARDS patients with composition changes exacerbates pulmonary endothelial dysfunction and acute lung injury induced by cecal ligation and puncture (CLP) in mice.Respir Res. 2020 Nov 4;21(1):293. doi: 10.1186/s12931-020-01553-3. Respir Res. 2020. PMID: 33148285 Free PMC article.
-
ISM1 suppresses LPS-induced acute lung injury and post-injury lung fibrosis in mice.Mol Med. 2022 Jun 25;28(1):72. doi: 10.1186/s10020-022-00500-w. Mol Med. 2022. PMID: 35752760 Free PMC article.
-
Delivery systems of therapeutic nucleic acids for the treatment of acute lung injury/acute respiratory distress syndrome.J Control Release. 2023 Aug;360:1-14. doi: 10.1016/j.jconrel.2023.06.018. Epub 2023 Jun 19. J Control Release. 2023. PMID: 37330013 Review.
-
Classic Signaling Pathways in Alveolar Injury and Repair Involved in Sepsis-Induced ALI/ARDS: New Research Progress and Prospect.Dis Markers. 2022 Jun 18;2022:6362344. doi: 10.1155/2022/6362344. eCollection 2022. Dis Markers. 2022. PMID: 35726235 Free PMC article. Review.
Cited by
-
SARS-CoV-2 ORF3a positively regulates NF-κB activity by enhancing IKKβ-NEMO interaction.Virus Res. 2023 Apr 15;328:199086. doi: 10.1016/j.virusres.2023.199086. Epub 2023 Mar 13. Virus Res. 2023. PMID: 36894068 Free PMC article.
-
Molecular mechanisms of Sepsis attacking the immune system and solid organs.Front Med (Lausanne). 2024 Aug 29;11:1429370. doi: 10.3389/fmed.2024.1429370. eCollection 2024. Front Med (Lausanne). 2024. PMID: 39267971 Free PMC article. Review.
-
Exploratory analysis to predict pneumonitis during durvalumab consolidation therapy for patients with locally advanced non-small cell lung cancer from proteomic profiling of circulating extracellular vesicles.Thorac Cancer. 2023 Oct;14(29):2909-2923. doi: 10.1111/1759-7714.15077. Epub 2023 Aug 24. Thorac Cancer. 2023. PMID: 37614219 Free PMC article.
-
miR-328-3p targets TLR2 to ameliorate oxygen-glucose deprivation injury and neutrophil extracellular trap formation in HUVECs via inhibition of the NF-κB signaling pathway.PLoS One. 2024 Feb 23;19(2):e0299382. doi: 10.1371/journal.pone.0299382. eCollection 2024. PLoS One. 2024. PMID: 38394259 Free PMC article.
-
Baicalin relieves complement alternative pathway activation-induced lung inflammation through inhibition of NF-κB pathway.BMC Complement Med Ther. 2024 Sep 13;24(1):334. doi: 10.1186/s12906-024-04622-y. BMC Complement Med Ther. 2024. PMID: 39272057 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
