

Epigenetic control of chromosome-associated lncRNA genes essential for replication and stability
- PMID: 36273230
- PMCID: PMC9588035
- DOI: 10.1038/s41467-022-34099-7
Epigenetic control of chromosome-associated lncRNA genes essential for replication and stability
Abstract
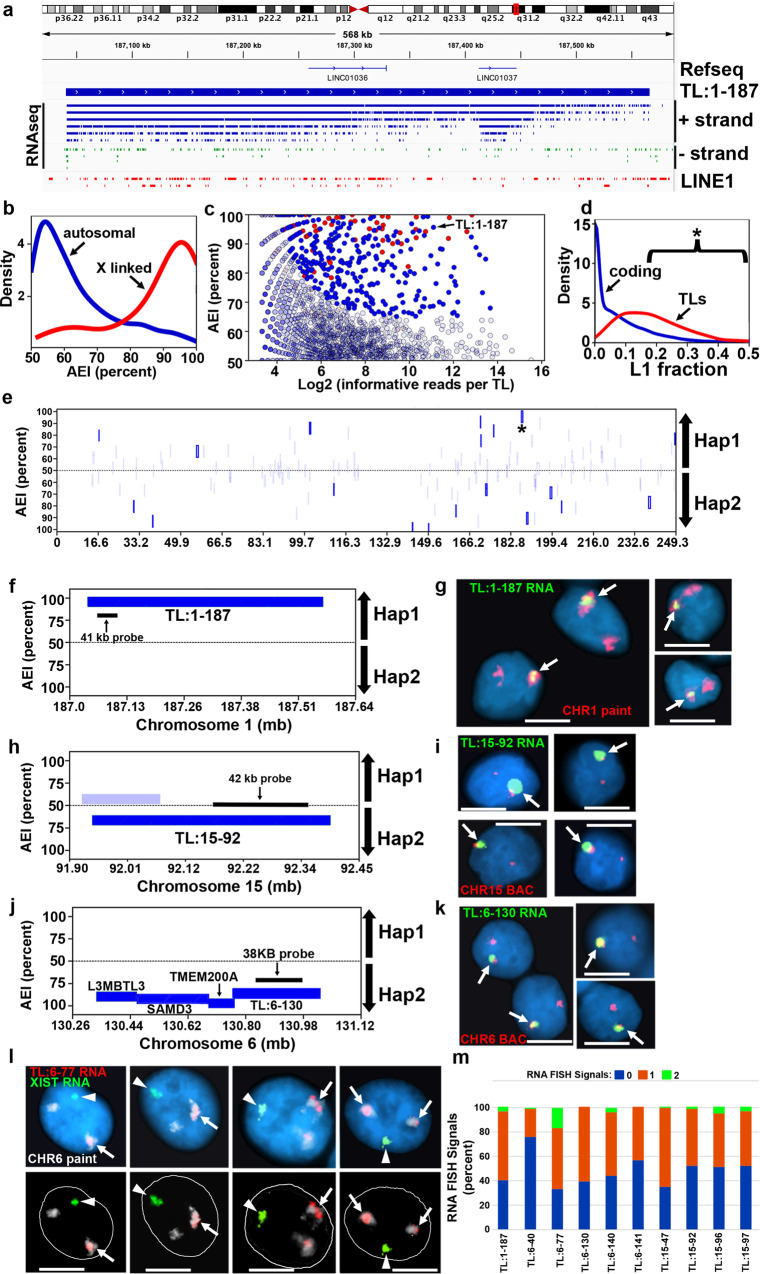
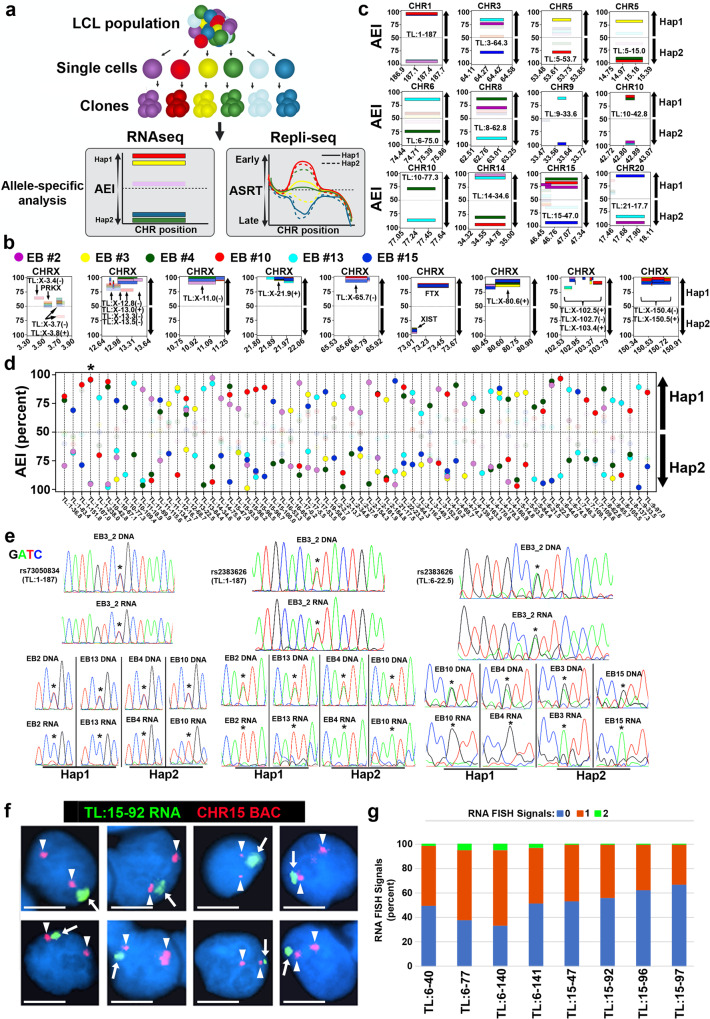
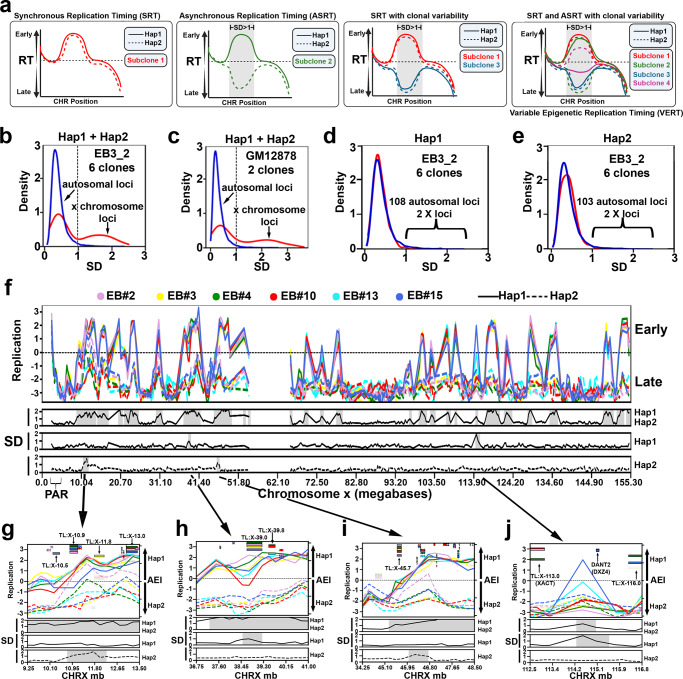
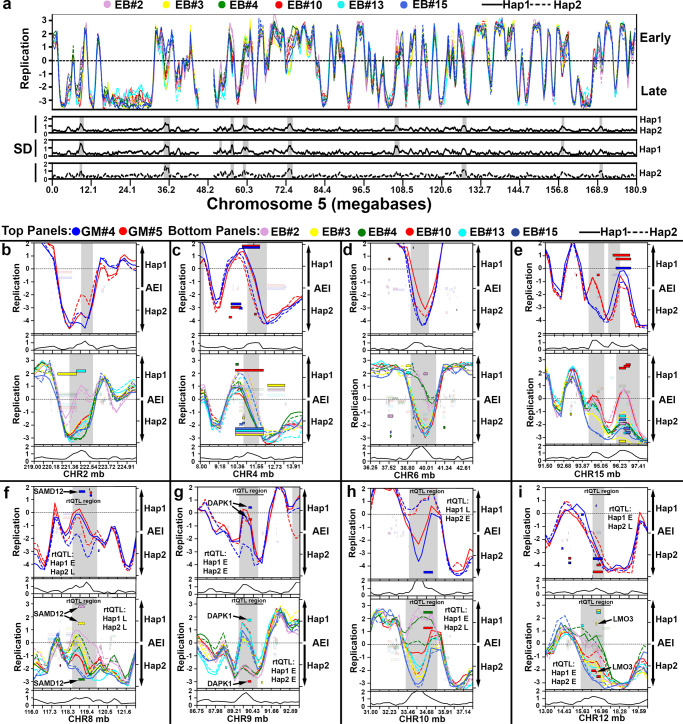
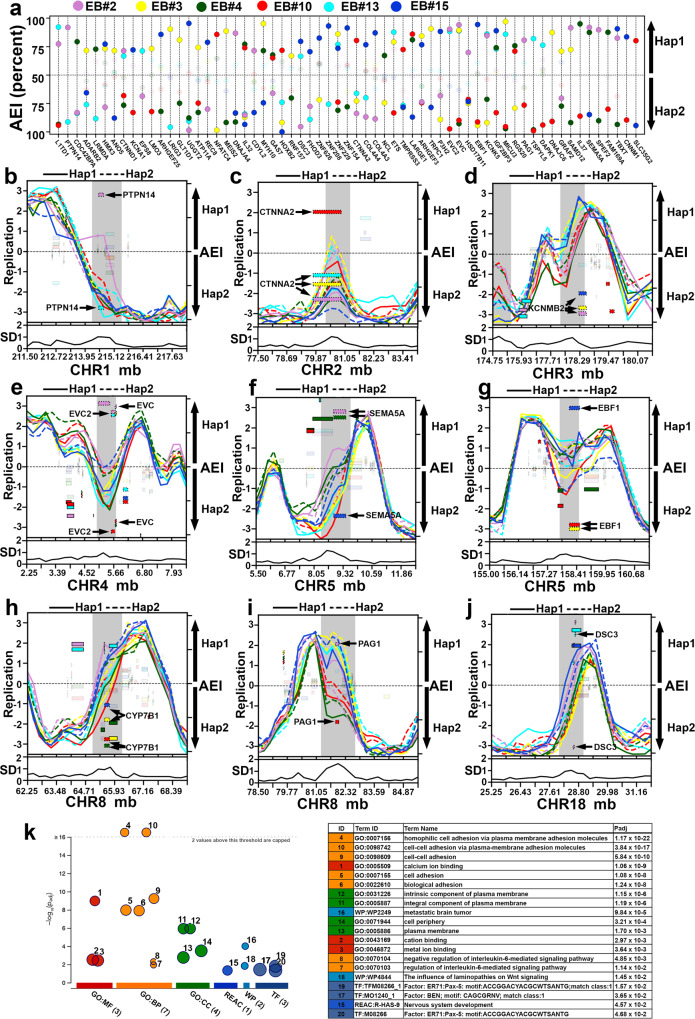
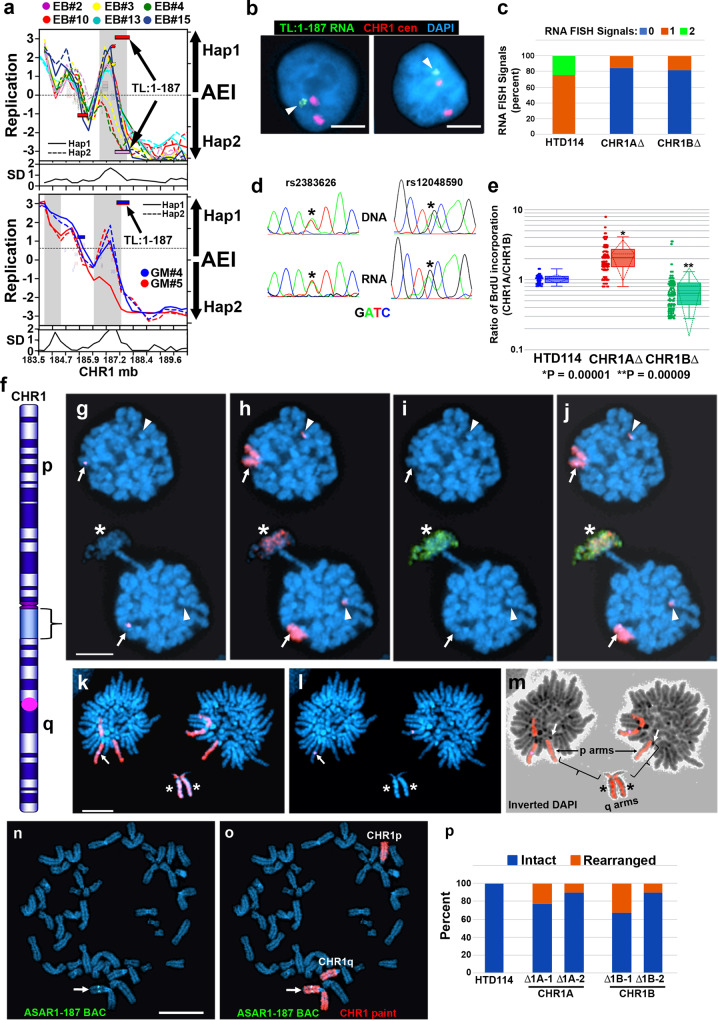
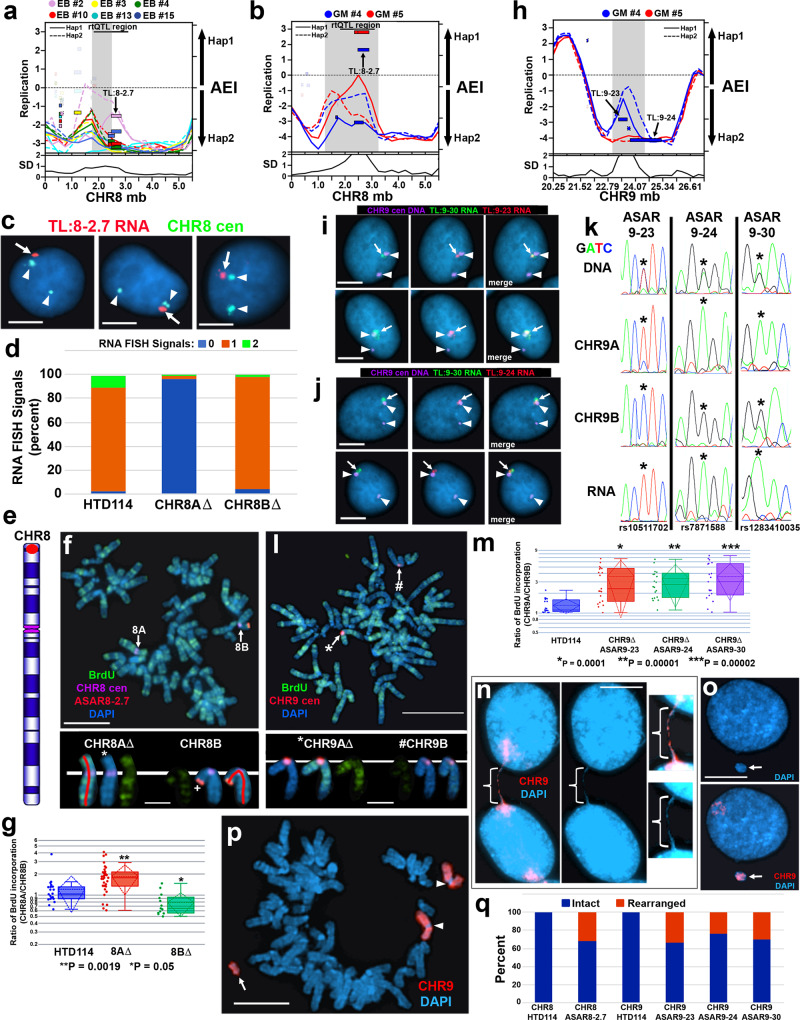
ASARs are long noncoding RNA genes that control replication timing of entire human chromosomes in cis. The three known ASAR genes are located on human chromosomes 6 and 15, and are essential for chromosome integrity. To identify ASARs on all human chromosomes we utilize a set of distinctive ASAR characteristics that allow for the identification of hundreds of autosomal loci with epigenetically controlled, allele-restricted behavior in expression and replication timing of coding and noncoding genes, and is distinct from genomic imprinting. Disruption of noncoding RNA genes at five of five tested loci result in chromosome-wide delayed replication and chromosomal instability, validating their ASAR activity. In addition to the three known essential cis-acting chromosomal loci, origins, centromeres, and telomeres, we propose that all mammalian chromosomes also contain "Inactivation/Stability Centers" that display allele-restricted epigenetic regulation of protein coding and noncoding ASAR genes that are essential for replication and stability of each chromosome.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
ASAR lncRNAs control DNA replication timing through interactions with multiple hnRNP/RNA binding proteins.Elife. 2024 Jun 18;13:RP95898. doi: 10.7554/eLife.95898. Elife. 2024. PMID: 38896448 Free PMC article.
-
Reciprocal monoallelic expression of ASAR lncRNA genes controls replication timing of human chromosome 6.RNA. 2020 Jun;26(6):724-738. doi: 10.1261/rna.073114.119. Epub 2020 Mar 6. RNA. 2020. PMID: 32144193 Free PMC article.
-
Mammalian chromosomes contain cis-acting elements that control replication timing, mitotic condensation, and stability of entire chromosomes.Bioessays. 2012 Sep;34(9):760-70. doi: 10.1002/bies.201200035. Epub 2012 Jun 18. Bioessays. 2012. PMID: 22706734 Free PMC article. Review.
-
ASAR15, A cis-acting locus that controls chromosome-wide replication timing and stability of human chromosome 15.PLoS Genet. 2015 Jan 8;11(1):e1004923. doi: 10.1371/journal.pgen.1004923. eCollection 2015 Jan. PLoS Genet. 2015. PMID: 25569254 Free PMC article.
-
Genomic imprinting mechanisms in mammals.Mutat Res. 2008 Dec 1;647(1-2):77-85. doi: 10.1016/j.mrfmmm.2008.08.008. Epub 2008 Aug 20. Mutat Res. 2008. PMID: 18778719 Free PMC article. Review.
Cited by
-
Monoallelically expressed noncoding RNAs form nucleolar territories on NOR-containing chromosomes and regulate rRNA expression.Elife. 2024 Jan 19;13:e80684. doi: 10.7554/eLife.80684. Elife. 2024. PMID: 38240312 Free PMC article.
-
Lessons from discovery of true ADAR RNA editing sites in a human cell line.BMC Biol. 2023 Jul 19;21(1):160. doi: 10.1186/s12915-023-01651-w. BMC Biol. 2023. PMID: 37468903 Free PMC article.
-
Replication timing and transcriptional control: beyond cause and effect - part IV.Curr Opin Genet Dev. 2023 Apr;79:102031. doi: 10.1016/j.gde.2023.102031. Epub 2023 Mar 9. Curr Opin Genet Dev. 2023. PMID: 36905782 Free PMC article. Review.
-
Identification of a lncRNA/circRNA-miRNA-mRNA network in Nasopharyngeal Carcinoma by deep sequencing and bioinformatics analysis.J Cancer. 2024 Feb 11;15(7):1916-1928. doi: 10.7150/jca.91546. eCollection 2024. J Cancer. 2024. PMID: 38434987 Free PMC article.
-
ASAR lncRNAs control DNA replication timing through interactions with multiple hnRNP/RNA binding proteins.Elife. 2024 Jun 18;13:RP95898. doi: 10.7554/eLife.95898. Elife. 2024. PMID: 38896448 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
