

Semi-automated assembly of high-quality diploid human reference genomes
- PMID: 36261518
- PMCID: PMC9668749
- DOI: 10.1038/s41586-022-05325-5
Semi-automated assembly of high-quality diploid human reference genomes
Abstract
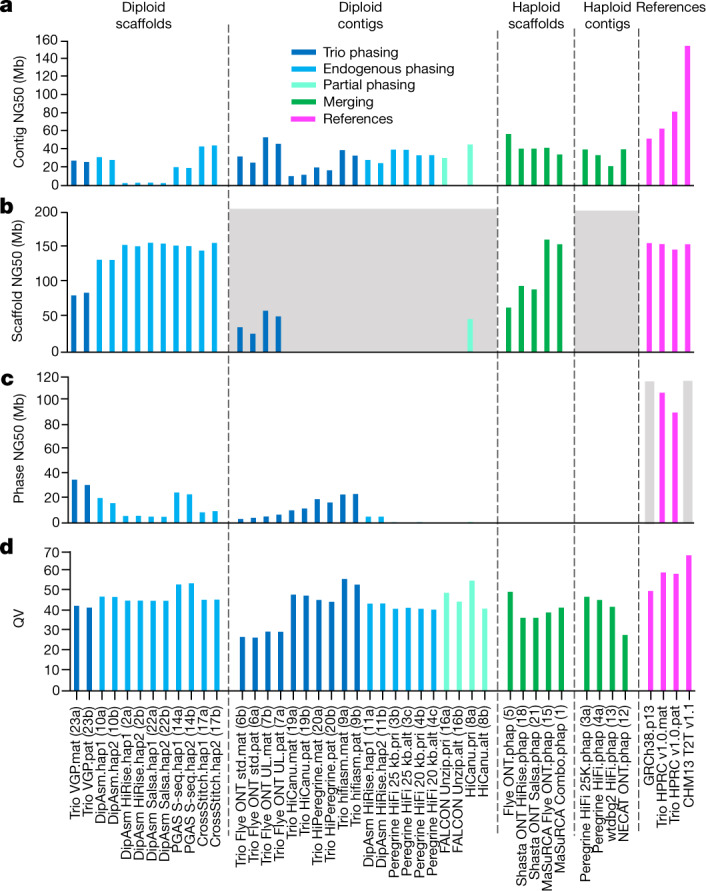
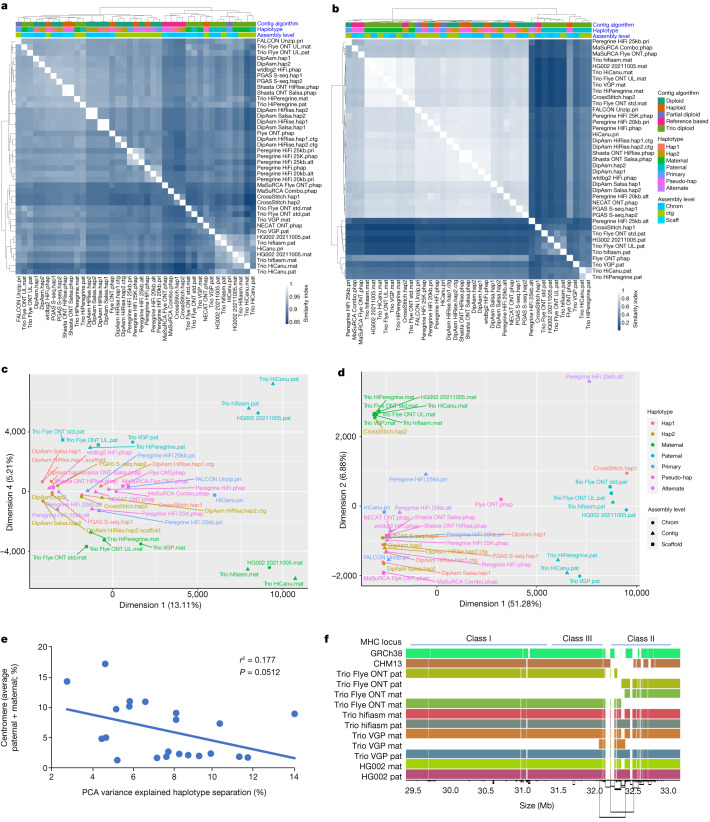
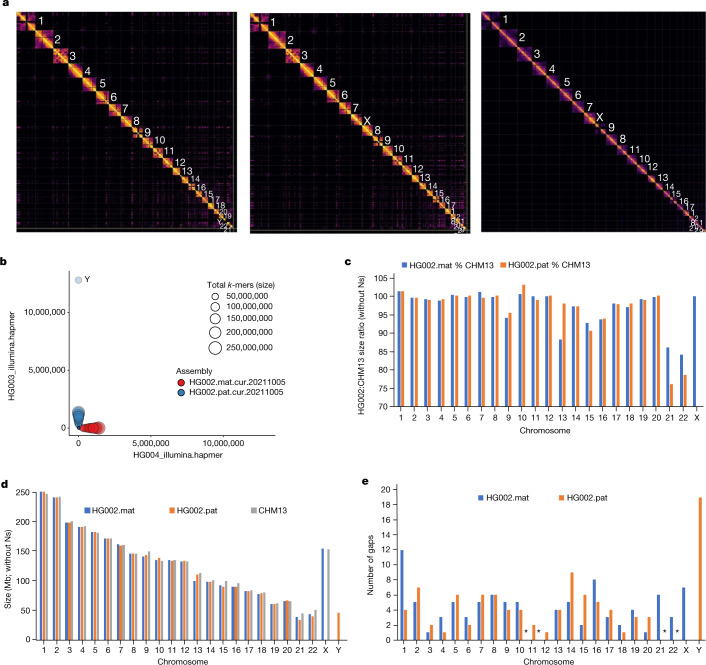
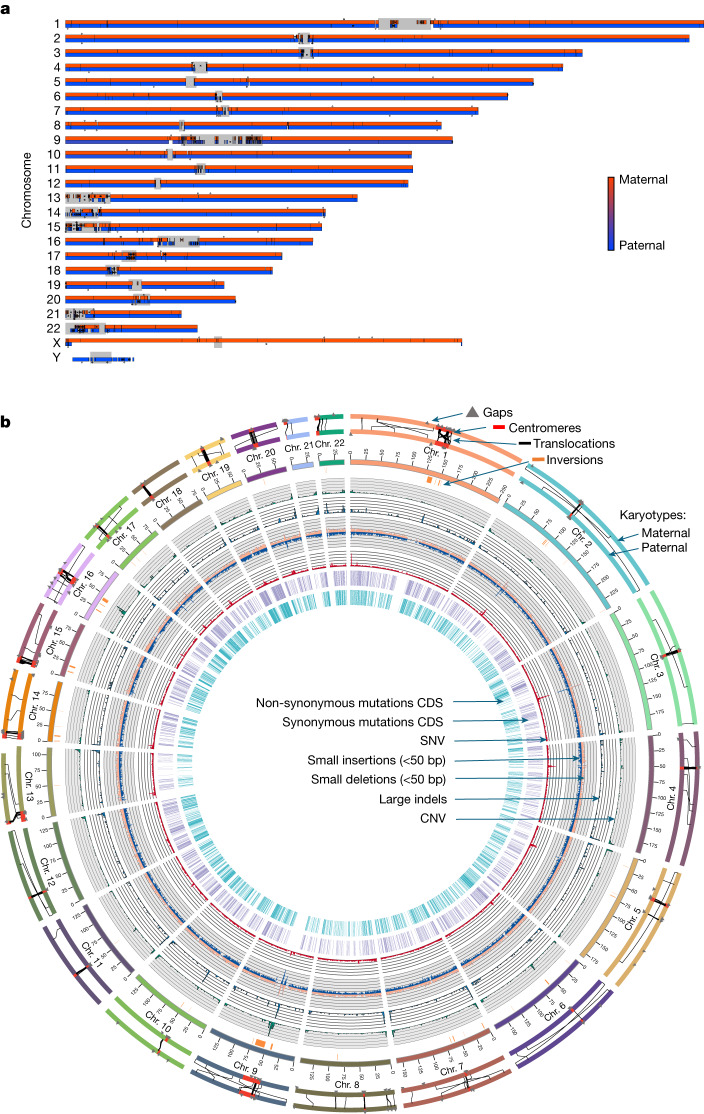
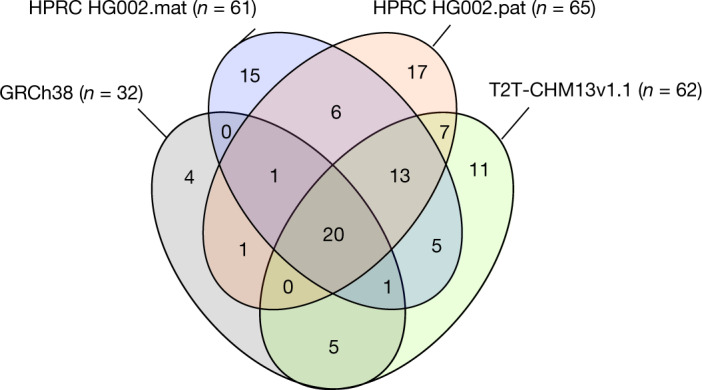
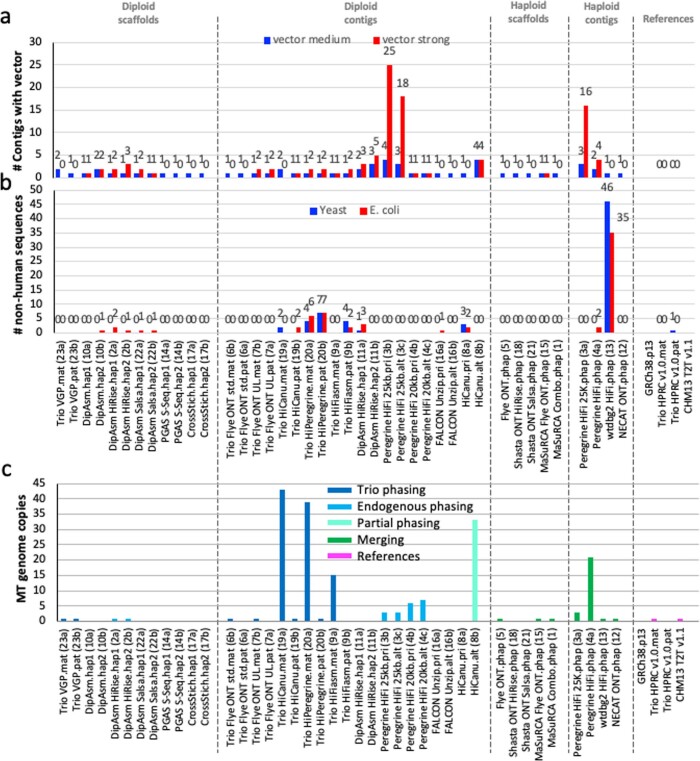
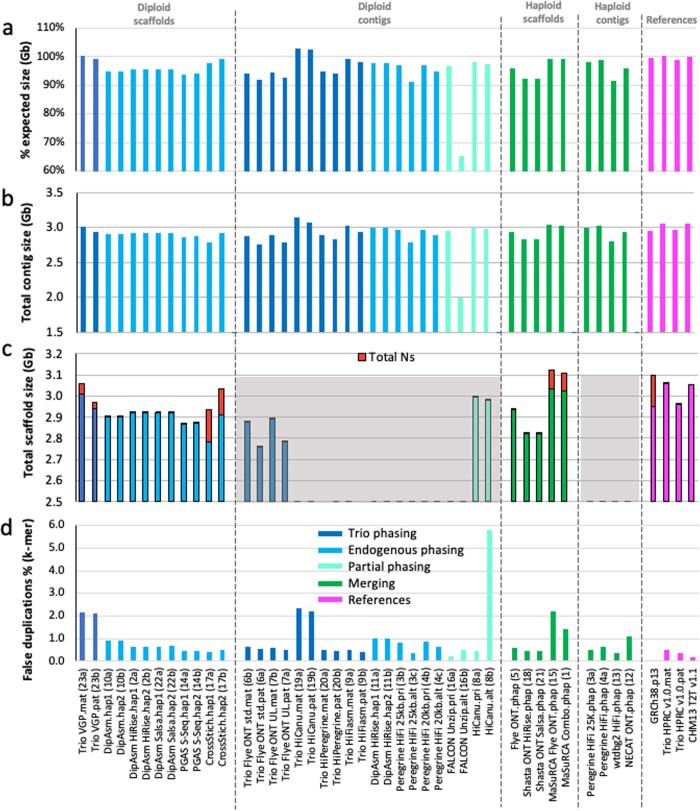
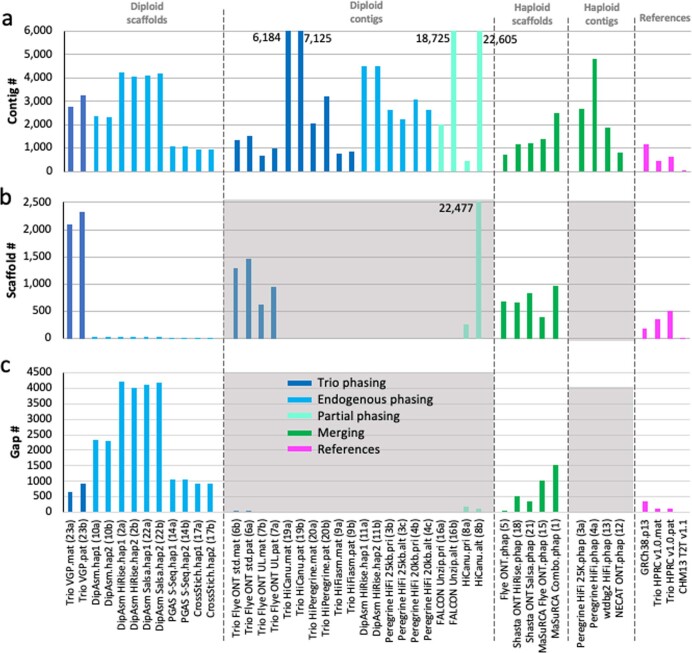
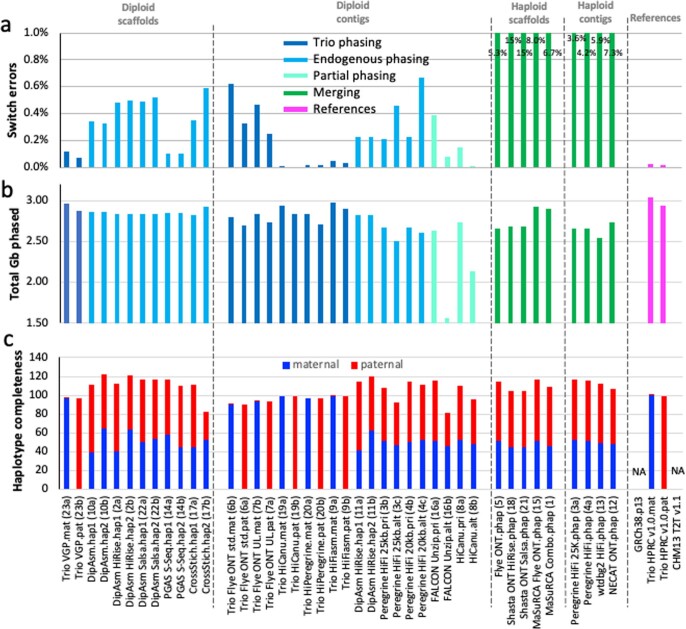
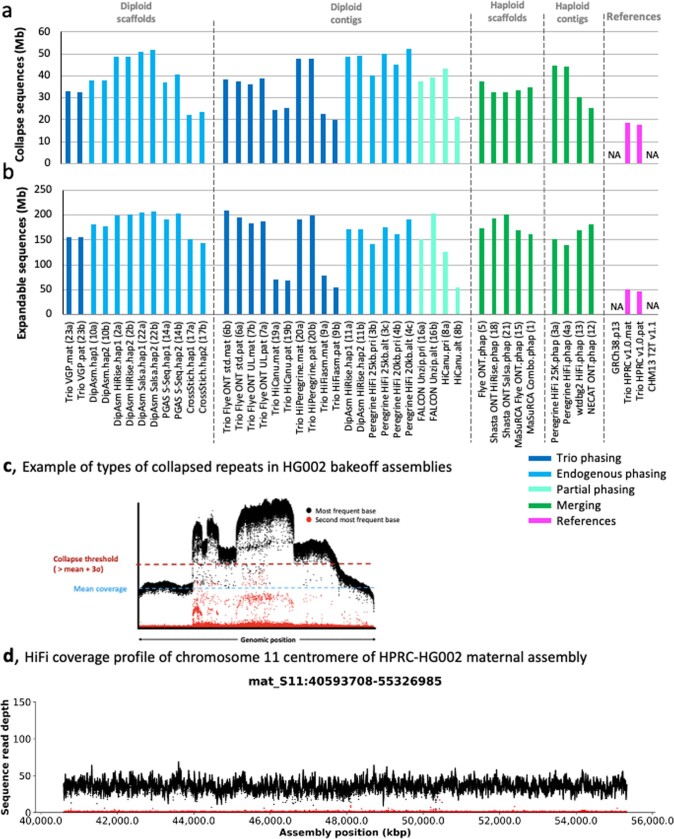
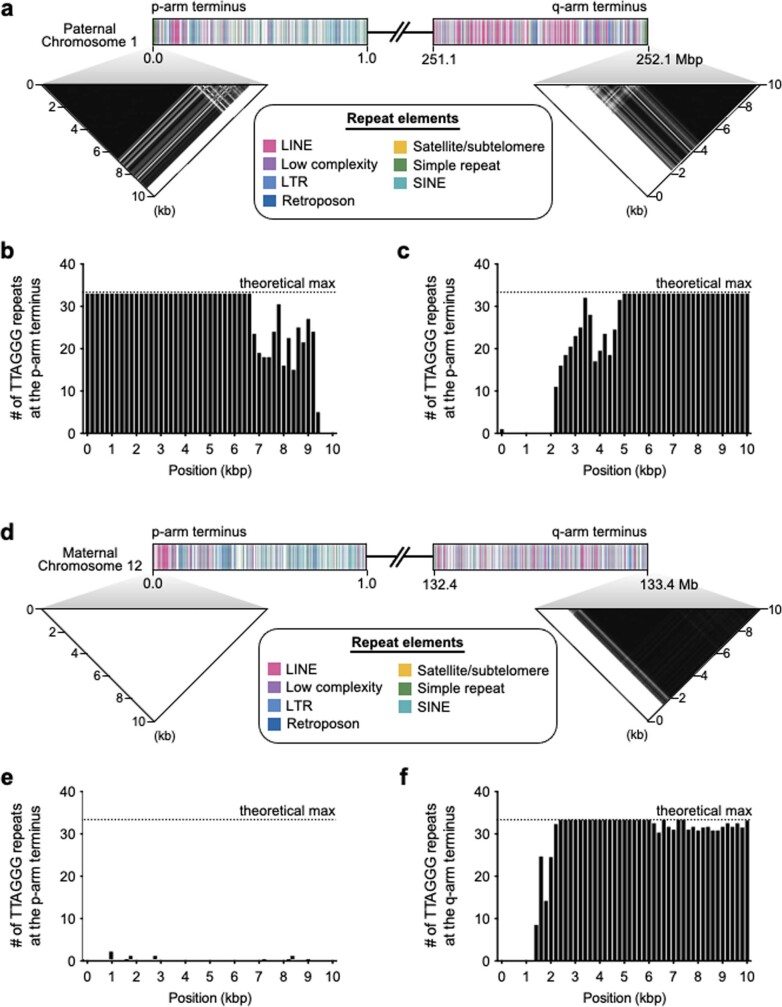
The current human reference genome, GRCh38, represents over 20 years of effort to generate a high-quality assembly, which has benefitted society1,2. However, it still has many gaps and errors, and does not represent a biological genome as it is a blend of multiple individuals3,4. Recently, a high-quality telomere-to-telomere reference, CHM13, was generated with the latest long-read technologies, but it was derived from a hydatidiform mole cell line with a nearly homozygous genome5. To address these limitations, the Human Pangenome Reference Consortium formed with the goal of creating high-quality, cost-effective, diploid genome assemblies for a pangenome reference that represents human genetic diversity6. Here, in our first scientific report, we determined which combination of current genome sequencing and assembly approaches yield the most complete and accurate diploid genome assembly with minimal manual curation. Approaches that used highly accurate long reads and parent-child data with graph-based haplotype phasing during assembly outperformed those that did not. Developing a combination of the top-performing methods, we generated our first high-quality diploid reference assembly, containing only approximately four gaps per chromosome on average, with most chromosomes within ±1% of the length of CHM13. Nearly 48% of protein-coding genes have non-synonymous amino acid changes between haplotypes, and centromeric regions showed the highest diversity. Our findings serve as a foundation for assembling near-complete diploid human genomes at scale for a pangenome reference to capture global genetic variation from single nucleotides to structural rearrangements.
© 2022. The Author(s).
Conflict of interest statement
E.E.E. was a scientific advisory board member of Variant Bio, Inc. J.K. and A.W. were full-time employees at Pacific Biosciences, a company developing single-molecule sequencing technologies. A.H. and J. Lee. were employees of Bionano Genomics, a company developing optical maps for genome assembly. J.G. and R.E.G. were affiliated with Dovetail Genomics, a company developing genome assembly tools, including Hi-C. A. Granat and E.B.J. were employees of Ilumina, Inc., a genome company generating short reads. A. Schmitt and S.S. were employees of Arima Genomics, a company developing Hi-C data for genome assemblies. All other authors declare no competing interests.
Figures

Similar articles
-
De novo assembly and phasing of a Korean human genome.Nature. 2016 Oct 13;538(7624):243-247. doi: 10.1038/nature20098. Epub 2016 Oct 5. Nature. 2016. PMID: 27706134
-
A draft human pangenome reference.Nature. 2023 May;617(7960):312-324. doi: 10.1038/s41586-023-05896-x. Epub 2023 May 10. Nature. 2023. PMID: 37165242 Free PMC article.
-
Dense and accurate whole-chromosome haplotyping of individual genomes.Nat Commun. 2017 Nov 3;8(1):1293. doi: 10.1038/s41467-017-01389-4. Nat Commun. 2017. PMID: 29101320 Free PMC article.
-
Haplotyping-Assisted Diploid Assembly and Variant Detection with Linked Reads.Methods Mol Biol. 2023;2590:161-182. doi: 10.1007/978-1-0716-2819-5_11. Methods Mol Biol. 2023. PMID: 36335499 Review.
-
The Human Pangenome Project: a global resource to map genomic diversity.Nature. 2022 Apr;604(7906):437-446. doi: 10.1038/s41586-022-04601-8. Epub 2022 Apr 20. Nature. 2022. PMID: 35444317 Free PMC article. Review.
Cited by
-
Improved sequence mapping using a complete reference genome and lift-over.Nat Methods. 2024 Jan;21(1):41-49. doi: 10.1038/s41592-023-02069-6. Epub 2023 Nov 30. Nat Methods. 2024. PMID: 38036856 Free PMC article.
-
The Telomere-Telomerase System Is Detrimental to Health at High-Altitude.Int J Environ Res Public Health. 2023 Jan 20;20(3):1935. doi: 10.3390/ijerph20031935. Int J Environ Res Public Health. 2023. PMID: 36767300 Free PMC article.
-
Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation.bioRxiv [Preprint]. 2023 Apr 5:2023.01.12.523790. doi: 10.1101/2023.01.12.523790. bioRxiv. 2023. Update in: Nat Methods. 2023 Oct;20(10):1483-1492. doi: 10.1038/s41592-023-01993-x PMID: 36711673 Free PMC article. Updated. Preprint.
-
Coverage-preserving sparsification of overlap graphs for long-read assembly.Bioinformatics. 2023 Mar 1;39(3):btad124. doi: 10.1093/bioinformatics/btad124. Bioinformatics. 2023. PMID: 36892439 Free PMC article.
-
Comparing genomic and epigenomic features across species using the WashU Comparative Epigenome Browser.Genome Res. 2023 May;33(5):824-835. doi: 10.1101/gr.277550.122. Epub 2023 May 8. Genome Res. 2023. PMID: 37156621 Free PMC article.
References
-
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 HG006677/HG/NHGRI NIH HHS/United States
- U01 HG010961/HG/NHGRI NIH HHS/United States
- R35 GM130151/GM/NIGMS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 HG010169/HG/NHGRI NIH HHS/United States
- U01 CA253481/CA/NCI NIH HHS/United States
- R01 HG011274/HG/NHGRI NIH HHS/United States
- U01 HG010971/HG/NHGRI NIH HHS/United States
- K99 HG010906/HG/NHGRI NIH HHS/United States
- R01 HG002385/HG/NHGRI NIH HHS/United States
- R01 HG010040/HG/NHGRI NIH HHS/United States
- U41 HG010972/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
