

Druggable transcriptomic pathways revealed in Parkinson's patient-derived midbrain neurons
- PMID: 36258029
- PMCID: PMC9579158
- DOI: 10.1038/s41531-022-00400-0
Druggable transcriptomic pathways revealed in Parkinson's patient-derived midbrain neurons
Abstract
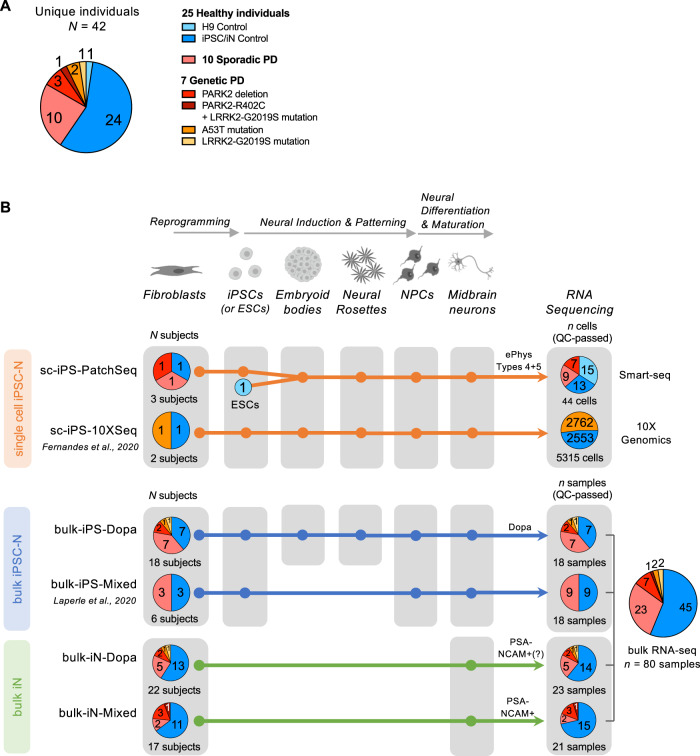
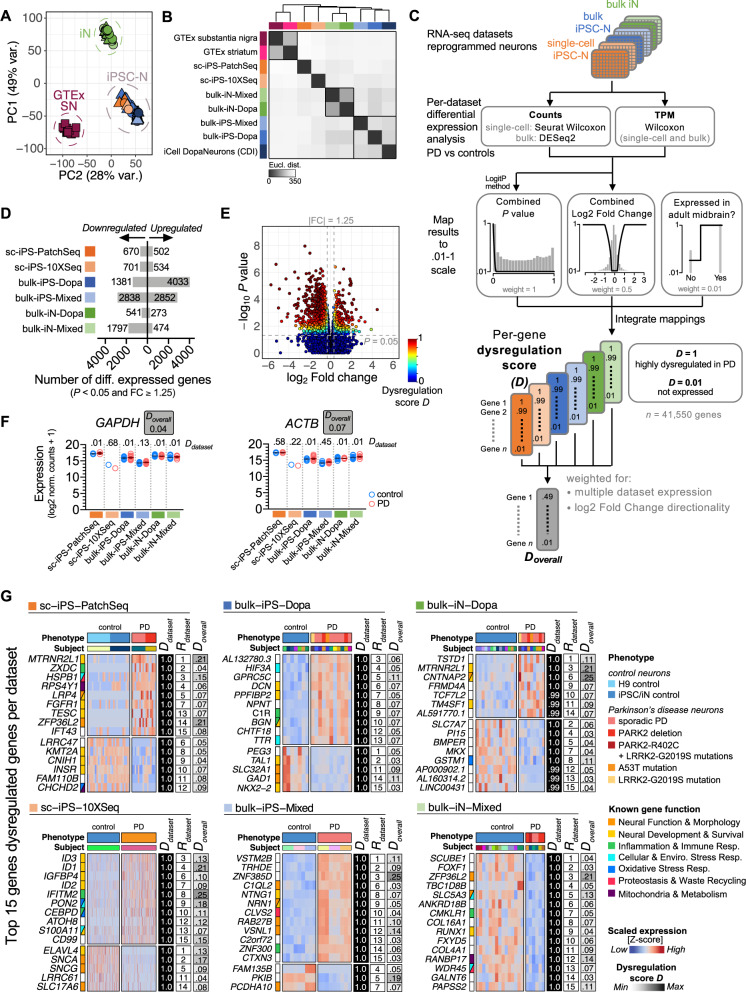
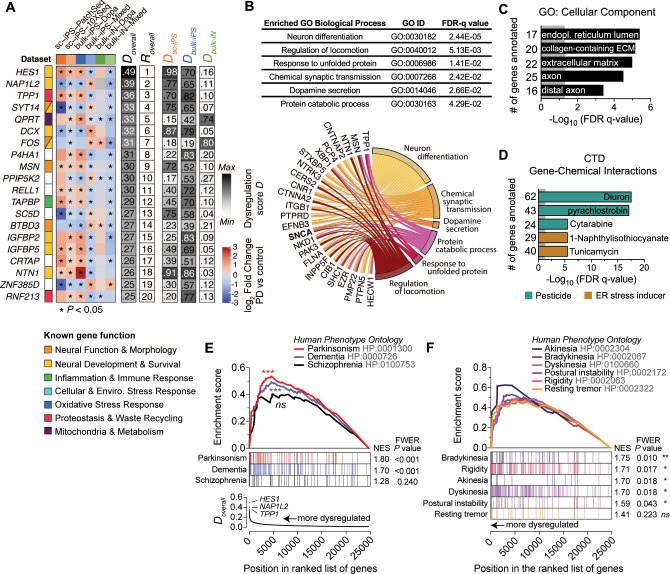
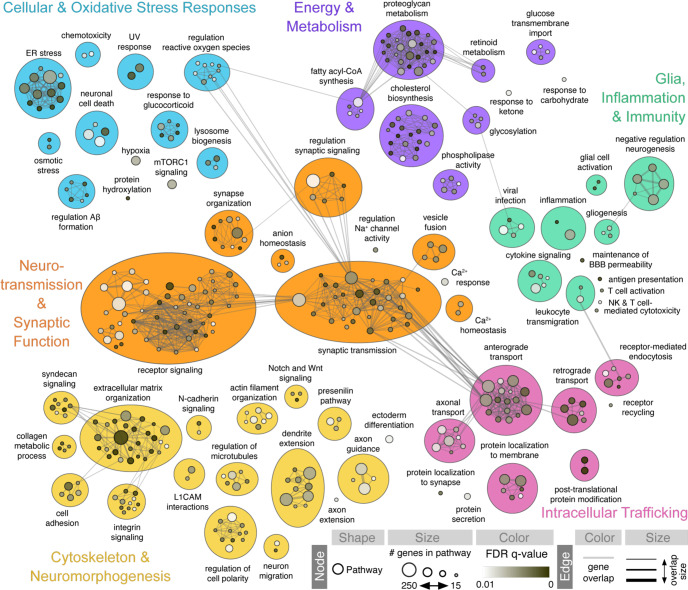
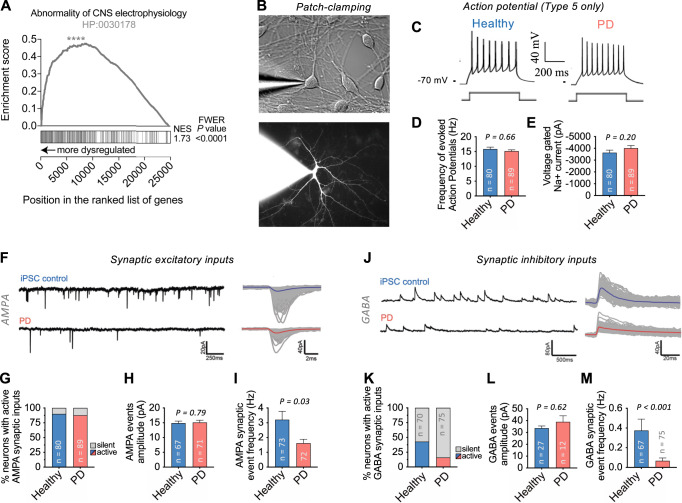
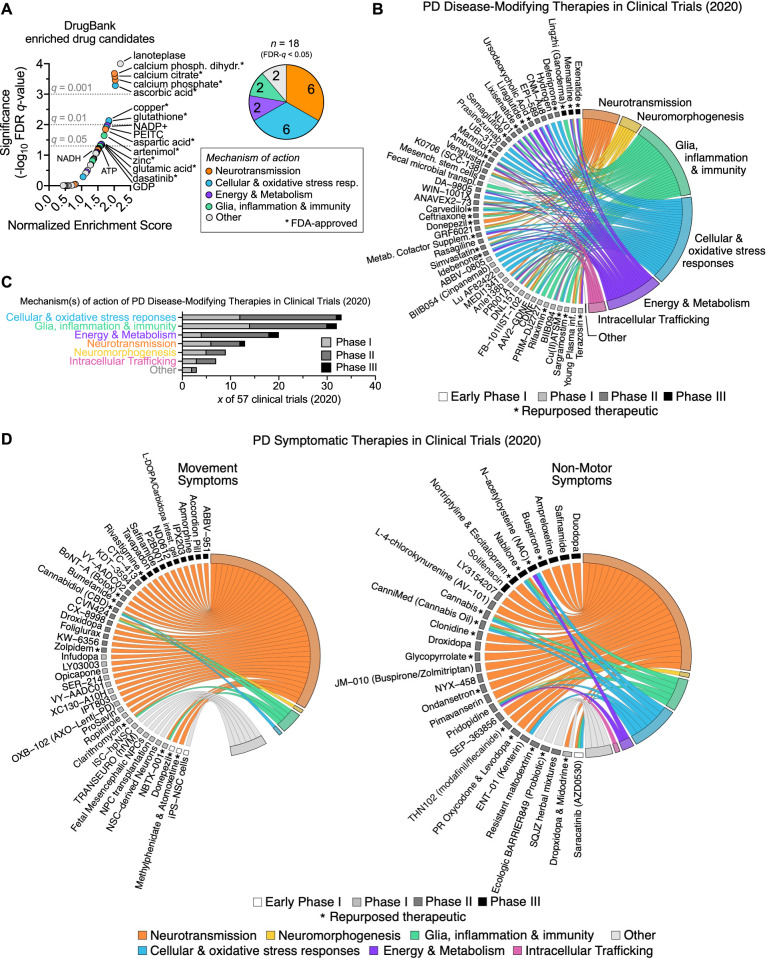
Complex genetic predispositions accelerate the chronic degeneration of midbrain substantia nigra neurons in Parkinson's disease (PD). Deciphering the human molecular makeup of PD pathophysiology can guide the discovery of therapeutics to slow the disease progression. However, insights from human postmortem brain studies only portray the latter stages of PD, and there is a lack of data surrounding molecular events preceding the neuronal loss in patients. We address this gap by identifying the gene dysregulation of live midbrain neurons reprogrammed in vitro from the skin cells of 42 individuals, including sporadic and familial PD patients and matched healthy controls. To minimize bias resulting from neuronal reprogramming and RNA-seq methods, we developed an analysis pipeline integrating PD transcriptomes from different RNA-seq datasets (unsorted and sorted bulk vs. single-cell and Patch-seq) and reprogramming strategies (induced pluripotency vs. direct conversion). This PD cohort's transcriptome is enriched for human genes associated with known clinical phenotypes of PD, regulation of locomotion, bradykinesia and rigidity. Dysregulated gene expression emerges strongest in pathways underlying synaptic transmission, metabolism, intracellular trafficking, neural morphogenesis and cellular stress/immune responses. We confirmed a synaptic impairment with patch-clamping and identified pesticides and endoplasmic reticulum stressors as the most significant gene-chemical interactions in PD. Subsequently, we associated the PD transcriptomic profile with candidate pharmaceuticals in a large database and a registry of current clinical trials. This study highlights human transcriptomic pathways that can be targeted therapeutically before the irreversible neuronal loss. Furthermore, it demonstrates the preclinical relevance of unbiased large transcriptomic assays of reprogrammed patient neurons.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
The landscape of multiscale transcriptomic networks and key regulators in Parkinson's disease.Nat Commun. 2019 Nov 20;10(1):5234. doi: 10.1038/s41467-019-13144-y. Nat Commun. 2019. PMID: 31748532 Free PMC article.
-
Reduced synaptic activity and dysregulated extracellular matrix pathways in midbrain neurons from Parkinson's disease patients.NPJ Parkinsons Dis. 2022 Aug 10;8(1):103. doi: 10.1038/s41531-022-00366-z. NPJ Parkinsons Dis. 2022. PMID: 35948563 Free PMC article.
-
Trib3 Is Elevated in Parkinson's Disease and Mediates Death in Parkinson's Disease Models.J Neurosci. 2015 Jul 29;35(30):10731-49. doi: 10.1523/JNEUROSCI.0614-15.2015. J Neurosci. 2015. PMID: 26224857 Free PMC article.
-
Genetic predispositions of Parkinson's disease revealed in patient-derived brain cells.NPJ Parkinsons Dis. 2020 Apr 24;6:8. doi: 10.1038/s41531-020-0110-8. eCollection 2020. NPJ Parkinsons Dis. 2020. PMID: 32352027 Free PMC article. Review.
-
Therapeutic potentials of plant iridoids in Alzheimer's and Parkinson's diseases: A review.Eur J Med Chem. 2019 May 1;169:185-199. doi: 10.1016/j.ejmech.2019.03.009. Epub 2019 Mar 8. Eur J Med Chem. 2019. PMID: 30877973 Review.
Cited by
-
Leveraging neighborhood representations of single-cell data to achieve sensitive DE testing with miloDE.Genome Biol. 2024 Jul 18;25(1):189. doi: 10.1186/s13059-024-03334-3. Genome Biol. 2024. PMID: 39026254 Free PMC article.
-
Multimodal Nature of the Single-cell Primate Brain Atlas: Morphology, Transcriptome, Electrophysiology, and Connectivity.Neurosci Bull. 2024 Apr;40(4):517-532. doi: 10.1007/s12264-023-01160-4. Epub 2024 Jan 9. Neurosci Bull. 2024. PMID: 38194157 Free PMC article. Review.
-
Focusing on the tetra-partite synapse in Parkinson's disease research using human patient-derived neurons.Neural Regen Res. 2024 May;19(5):979-981. doi: 10.4103/1673-5374.382235. Neural Regen Res. 2024. PMID: 37862197 Free PMC article. No abstract available.
-
Genomic insights into the comorbidity between type 2 diabetes and schizophrenia.Schizophrenia (Heidelb). 2024 Feb 21;10(1):22. doi: 10.1038/s41537-024-00445-5. Schizophrenia (Heidelb). 2024. PMID: 38383672 Free PMC article.
-
How is Excitotoxicity Being Modelled in iPSC-Derived Neurons?Neurotox Res. 2024 Oct 15;42(5):43. doi: 10.1007/s12640-024-00721-3. Neurotox Res. 2024. PMID: 39405005 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Miscellaneous
