

Herpes simplex virus type 2 inhibits TNF-α-induced NF-κB activation through viral protein ICP22-mediated interaction with p65
- PMID: 36211339
- PMCID: PMC9538160
- DOI: 10.3389/fimmu.2022.983502
Herpes simplex virus type 2 inhibits TNF-α-induced NF-κB activation through viral protein ICP22-mediated interaction with p65
Abstract
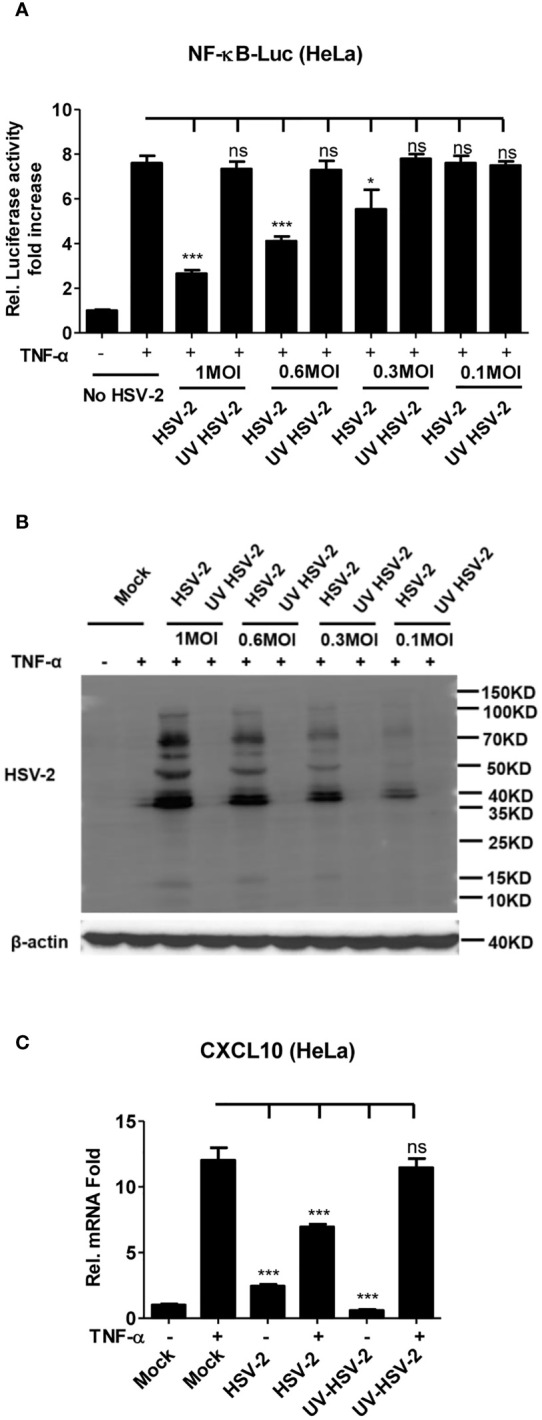
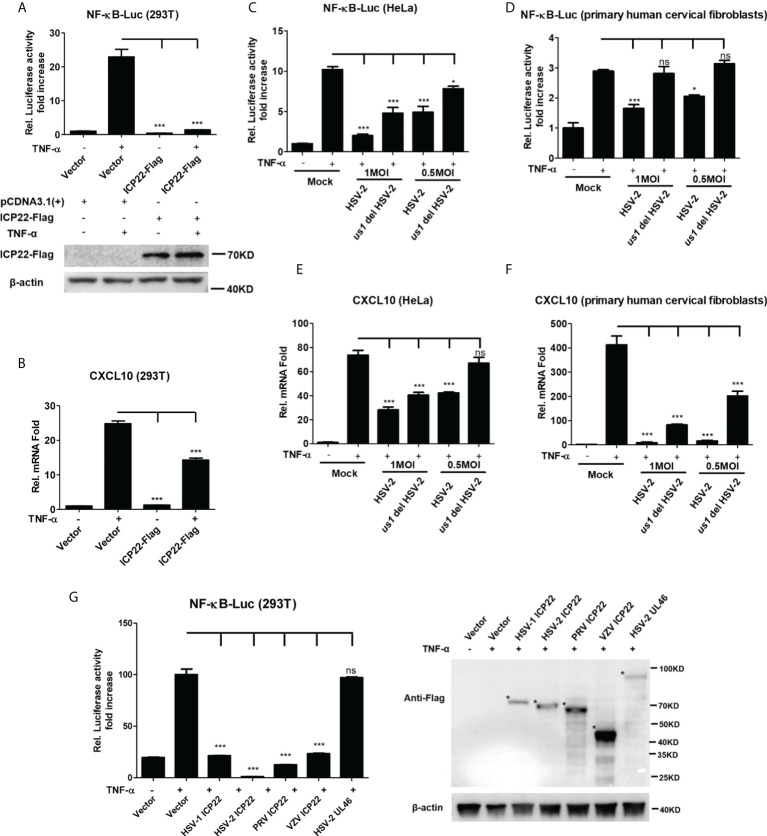
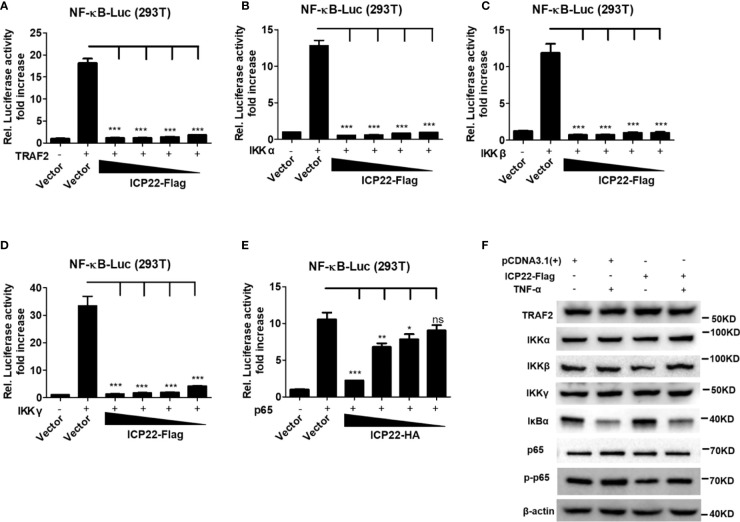
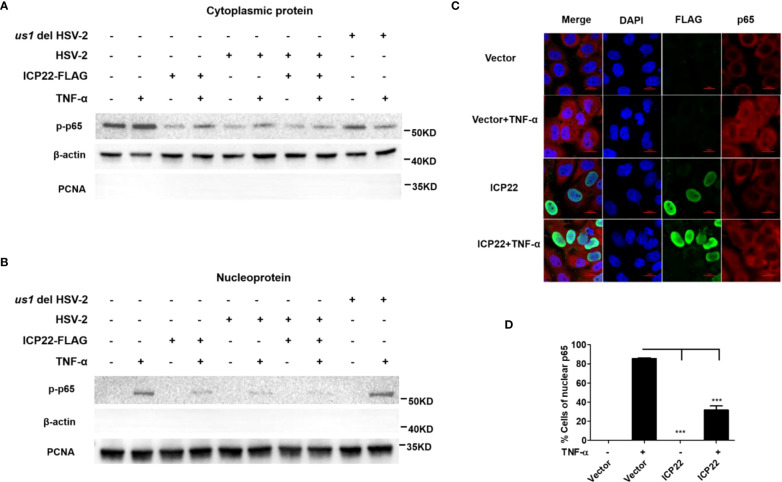
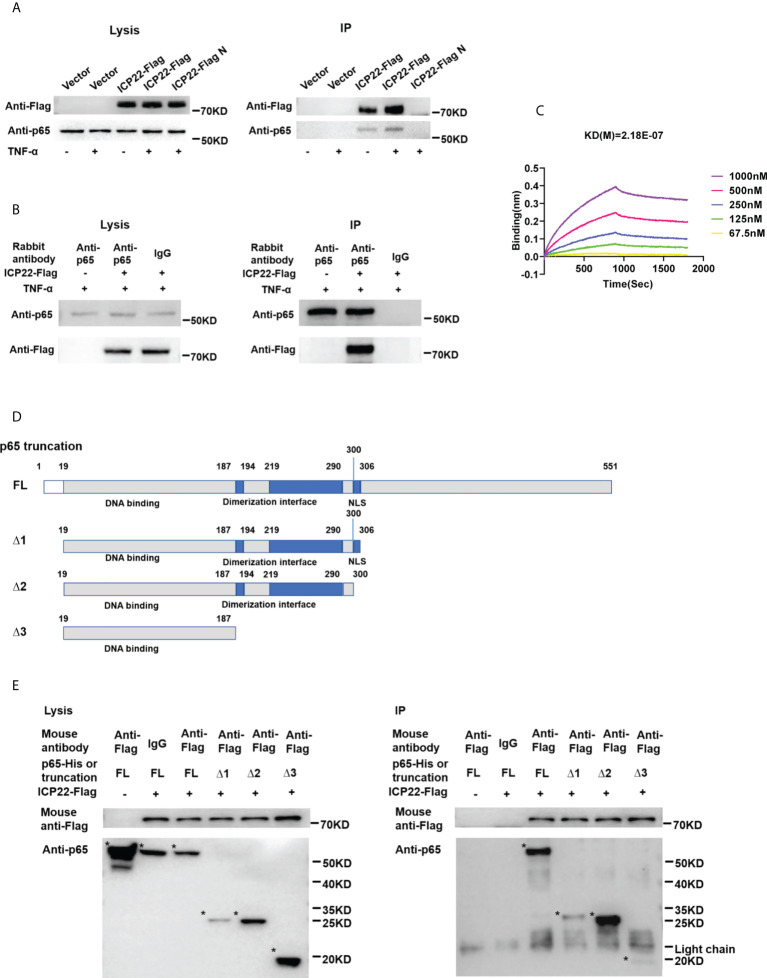
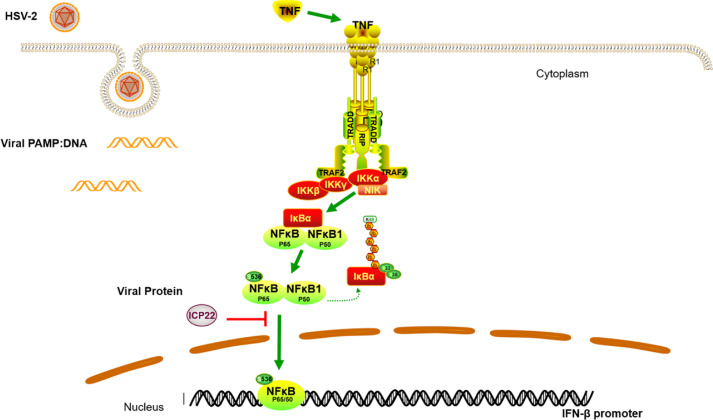
Herpes simplex virus type 2 (HSV-2) is a prevalent human pathogen and the main cause of genital herpes. After initial infection, HSV-2 can establish lifelong latency within dorsal root ganglia by evading the innate immunity of the host. NF-κB has a crucial role in regulating cell proliferation, inflammation, apoptosis, and immune responses. It is known that inhibition of NF-κB activation by a virus could facilitate it to establish infection in the host. In the current study, we found that HSV-2 inhibited TNF-α-induced activation of NF-κB-responsive promoter in a dose-dependent manner, while UV-inactivated HSV-2 did not have such capability. We further identified the immediate early protein ICP22 of HSV-2 as a vital viral element in inhibiting the activation of NF-κB-responsive promoter. The role of ICP22 was confirmed in human cervical cell line HeLa and primary cervical fibroblasts in the context of HSV-2 infection, showing that ICP22 deficient HSV-2 largely lost the capability in suppressing NF-κB activation. HSV-2 ICP22 was further shown to suppress the activity of TNF receptor-associated factor 2 (TRAF2)-, IκB kinase α (IKK α)-, IKK β-, IKK γ-, or p65-induced activation of NF-κB-responsive promoter. Mechanistically, HSV-2 ICP22 inhibited the phosphorylation and nuclear translocation of p65 by directly interacting with p65, resulting in the blockade of NF-κB activation. Furthermore, ICP22 from several alpha-herpesviruses could also inhibit NF-κB activation, suggesting the significance of ICP22 in herpesvirus immune evasion. Findings in this study highlight the importance of ICP22 in inhibiting NF-κB activation, revealing a novel mechanism by which HSV-2 evades the host antiviral responses.
Keywords: HSV-2; ICP22; NF-κB; immune evasion; p65.
Copyright © 2022 Hu, Fu, Li, Zhang, Li, Hu and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-κB activation by interacting with p65/RelA and p50/NF-κB1.J Virol. 2013 Dec;87(23):12935-48. doi: 10.1128/JVI.01952-13. Epub 2013 Sep 25. J Virol. 2013. PMID: 24067962 Free PMC article.
-
Herpes Simplex Virus 1 UL2 Inhibits the TNF-α-Mediated NF-κB Activity by Interacting With p65/p50.Front Immunol. 2020 May 13;11:549. doi: 10.3389/fimmu.2020.00549. eCollection 2020. Front Immunol. 2020. PMID: 32477319 Free PMC article.
-
Herpes Simplex Virus Type 2 Inhibits Type I IFN Signaling Mediated by the Novel E3 Ubiquitin Protein Ligase Activity of Viral Protein ICP22.J Immunol. 2020 Sep 1;205(5):1281-1292. doi: 10.4049/jimmunol.2000418. Epub 2020 Jul 22. J Immunol. 2020. PMID: 32699158
-
Herpes Simplex Virus Type 2 Glycoprotein D Inhibits NF-κB Activation by Interacting with p65.J Immunol. 2021 Jun 15;206(12):2852-2861. doi: 10.4049/jimmunol.2001336. Epub 2021 May 28. J Immunol. 2021. PMID: 34049972
-
Multifaceted Roles of ICP22/ORF63 Proteins in the Life Cycle of Human Herpesviruses.Front Microbiol. 2021 Jun 7;12:668461. doi: 10.3389/fmicb.2021.668461. eCollection 2021. Front Microbiol. 2021. PMID: 34163446 Free PMC article. Review.
Cited by
-
CRISPR-Cas9 screen of E3 ubiquitin ligases identifies TRAF2 and UHRF1 as regulators of HIV latency in primary human T cells.mBio. 2024 Apr 10;15(4):e0222223. doi: 10.1128/mbio.02222-23. Epub 2024 Feb 27. mBio. 2024. PMID: 38411080 Free PMC article.
References
-
- McQuillan G, Kruszon-Moran D, Flagg EW, Paulose-Ram R. Prevalence of herpes simplex virus type 1 and type 2 in persons aged 14-49: United states, 2015-2016. NCHS Data Brief (2018) 304):1–8. - PubMed
-
- Wang K, Kappel JD, Canders C, Davila WF, Sayre D, Chavez M, et al. . A herpes simplex virus 2 glycoprotein d mutant generated by bacterial artificial chromosome mutagenesis is severely impaired for infecting neuronal cells and infects only vero cells expressing exogenous hvem. J Virol (2012) 86(23):12891–902. doi: 10.1128/JVI.01055-12 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources