

Role of TET dioxygenases in the regulation of both normal and pathological hematopoiesis
- PMID: 36203205
- PMCID: PMC9540719
- DOI: 10.1186/s13046-022-02496-x
Role of TET dioxygenases in the regulation of both normal and pathological hematopoiesis
Abstract
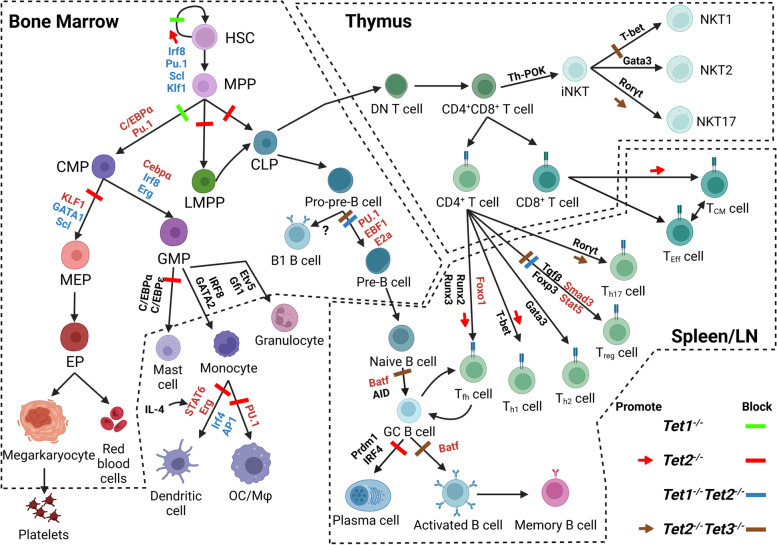
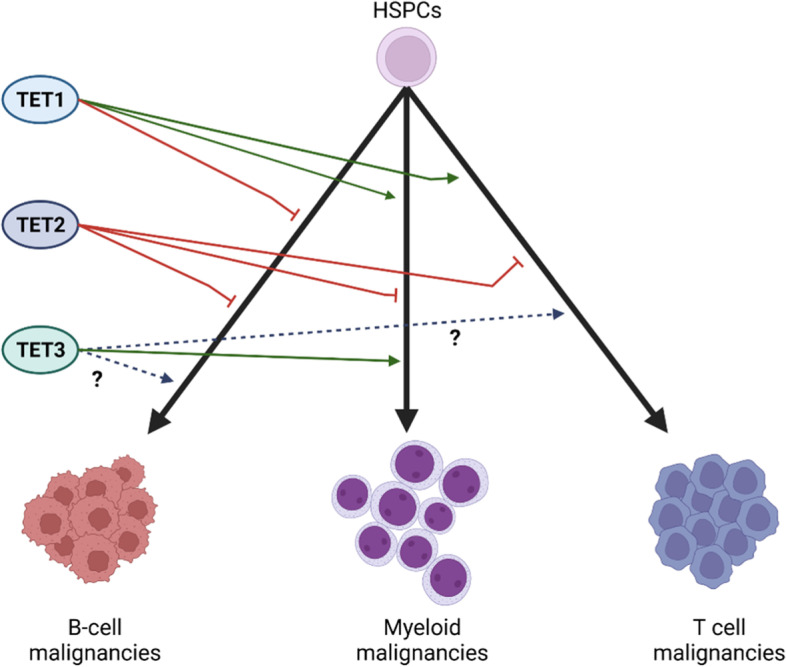
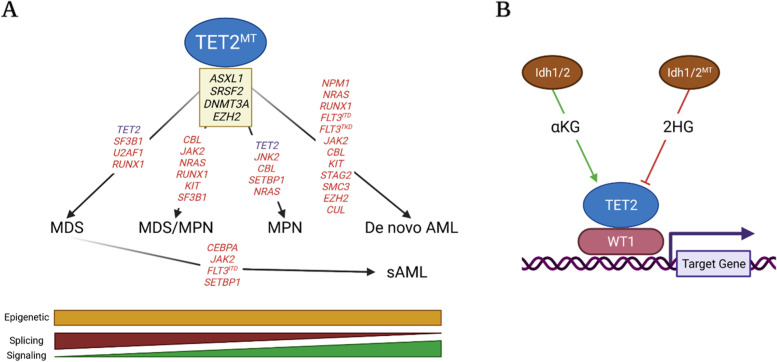
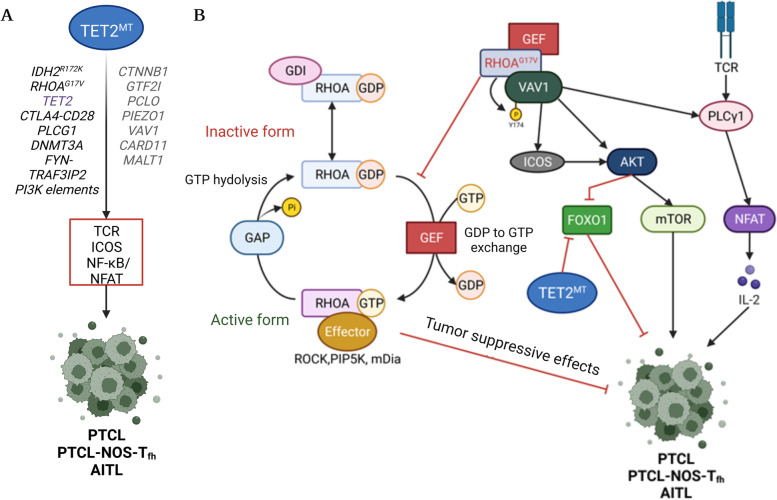
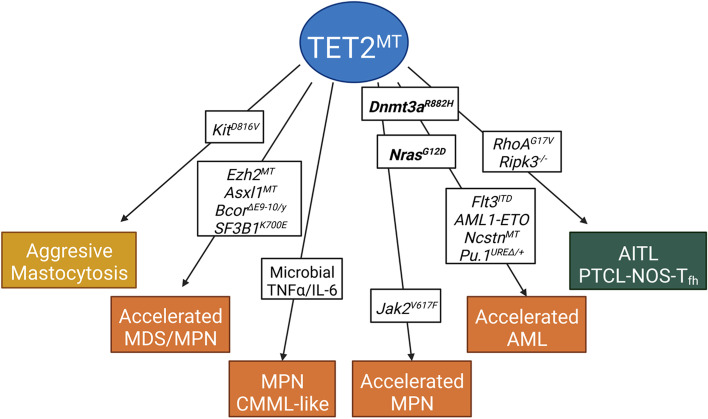
The family of ten-eleven translocation dioxygenases (TETs) consists of TET1, TET2, and TET3. Although all TETs are expressed in hematopoietic tissues, only TET2 is commonly found to be mutated in age-related clonal hematopoiesis and hematopoietic malignancies. TET2 mutation causes abnormal epigenetic landscape changes and results in multiple stages of lineage commitment/differentiation defects as well as genetic instability in hematopoietic stem/progenitor cells (HSPCs). TET2 mutations are founder mutations (first hits) in approximately 40-50% of cases of TET2-mutant (TET2MT) hematopoietic malignancies and are later hits in the remaining cases. In both situations, TET2MT collaborates with co-occurring mutations to promote malignant transformation. In TET2MT tumor cells, TET1 and TET3 partially compensate for TET2 activity and contribute to the pathogenesis of TET2MT hematopoietic malignancies. Here we summarize the most recent research on TETs in regulating of both normal and pathogenic hematopoiesis. We review the concomitant mutations and aberrant signals in TET2MT malignancies. We also discuss the molecular mechanisms by which concomitant mutations and aberrant signals determine lineage commitment in HSPCs and the identity of hematopoietic malignancies. Finally, we discuss potential strategies to treat TET2MT hematopoietic malignancies, including reverting the methylation state of TET2 target genes and targeting the concomitant mutations and aberrant signals.
Keywords: Concurring mutations; Differentiation; HSPCs; Leukemia; MDS; Self-renewal; TET2.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing financial or professional interests.
Figures
Similar articles
-
TET2 mutation as prototypic clonal hematopoiesis lesion.Semin Hematol. 2024 Feb;61(1):51-60. doi: 10.1053/j.seminhematol.2024.01.013. Epub 2024 Feb 2. Semin Hematol. 2024. PMID: 38431463 Free PMC article. Review.
-
TET proteins and 5-methylcytosine oxidation in hematological cancers.Immunol Rev. 2015 Jan;263(1):6-21. doi: 10.1111/imr.12239. Immunol Rev. 2015. PMID: 25510268 Free PMC article. Review.
-
Ten-Eleven-Translocation Genes in Cancer.Cancer Treat Res. 2023;190:363-373. doi: 10.1007/978-3-031-45654-1_11. Cancer Treat Res. 2023. PMID: 38113007 Review.
-
The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases.Leukemia. 2014 Mar;28(3):485-96. doi: 10.1038/leu.2013.337. Epub 2013 Nov 13. Leukemia. 2014. PMID: 24220273 Review.
-
Dysregulation of TET2 in hematologic malignancies.Int J Hematol. 2017 Jan;105(1):17-22. doi: 10.1007/s12185-016-2122-z. Epub 2016 Nov 15. Int J Hematol. 2017. PMID: 27848178 Review.
Cited by
-
Emerging DNA Methylome Targets in FLT3-ITD-Positive Acute Myeloid Leukemia: Combination Therapy with Clinically Approved FLT3 Inhibitors.Curr Treat Options Oncol. 2024 Jun;25(6):719-751. doi: 10.1007/s11864-024-01202-7. Epub 2024 May 2. Curr Treat Options Oncol. 2024. PMID: 38696033 Free PMC article. Review.
-
Aging is associated with functional and molecular changes in distinct hematopoietic stem cell subsets.Nat Commun. 2024 Sep 11;15(1):7966. doi: 10.1038/s41467-024-52318-1. Nat Commun. 2024. PMID: 39261515 Free PMC article.
-
TET2 mutation as prototypic clonal hematopoiesis lesion.Semin Hematol. 2024 Feb;61(1):51-60. doi: 10.1053/j.seminhematol.2024.01.013. Epub 2024 Feb 2. Semin Hematol. 2024. PMID: 38431463 Free PMC article. Review.
-
Cell of origin epigenetic priming determines susceptibility to Tet2 mutation.Nat Commun. 2024 May 21;15(1):4325. doi: 10.1038/s41467-024-48508-6. Nat Commun. 2024. PMID: 38773071 Free PMC article.
-
Ten-Eleven Translocation 1 and 2 Enzymes Affect Human Skin Fibroblasts in an Age-Related Manner.Biomedicines. 2023 Jun 7;11(6):1659. doi: 10.3390/biomedicines11061659. Biomedicines. 2023. PMID: 37371754 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
