

Signaling pathways and targeted therapies in lung squamous cell carcinoma: mechanisms and clinical trials
- PMID: 36198685
- PMCID: PMC9535022
- DOI: 10.1038/s41392-022-01200-x
Signaling pathways and targeted therapies in lung squamous cell carcinoma: mechanisms and clinical trials
Abstract
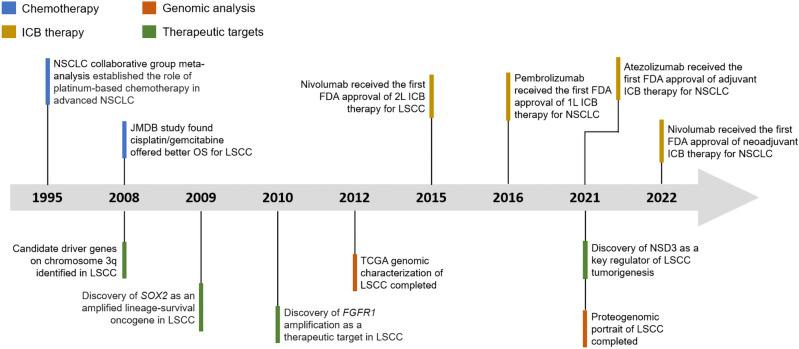
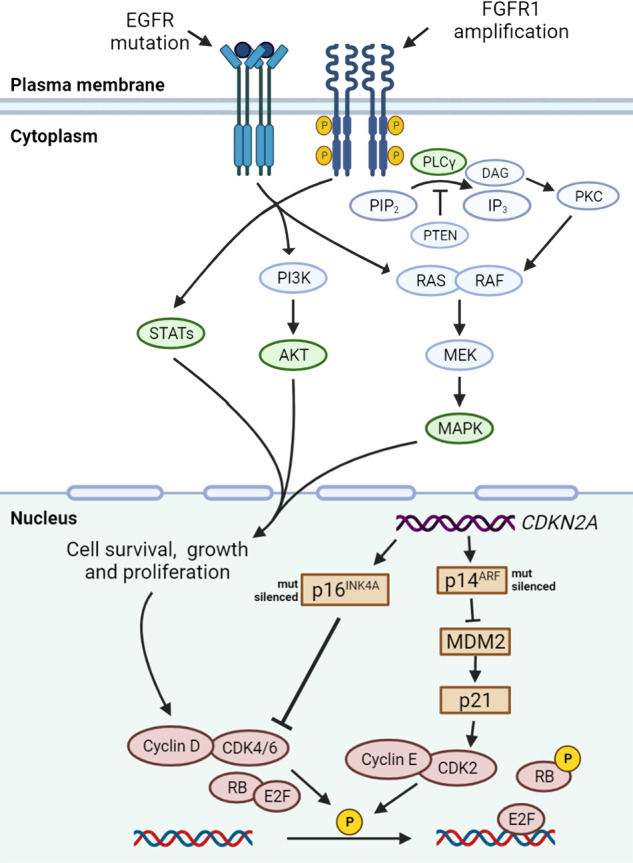
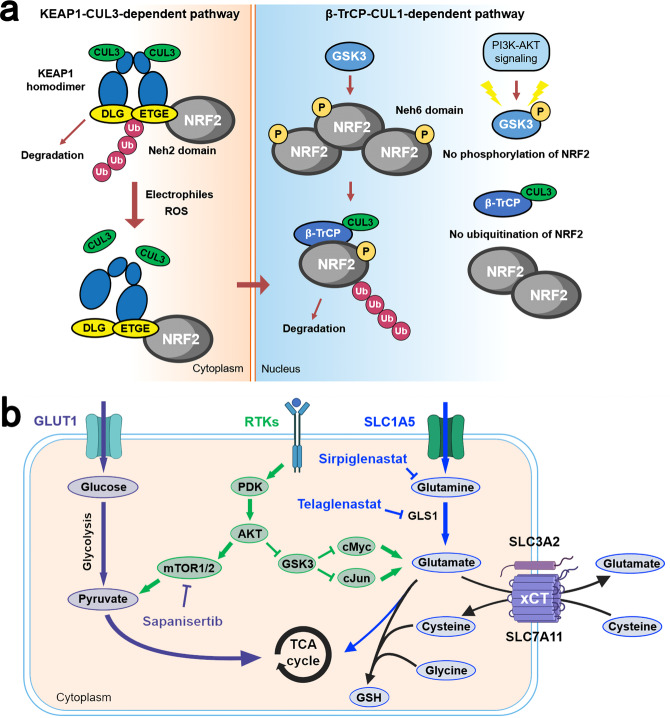
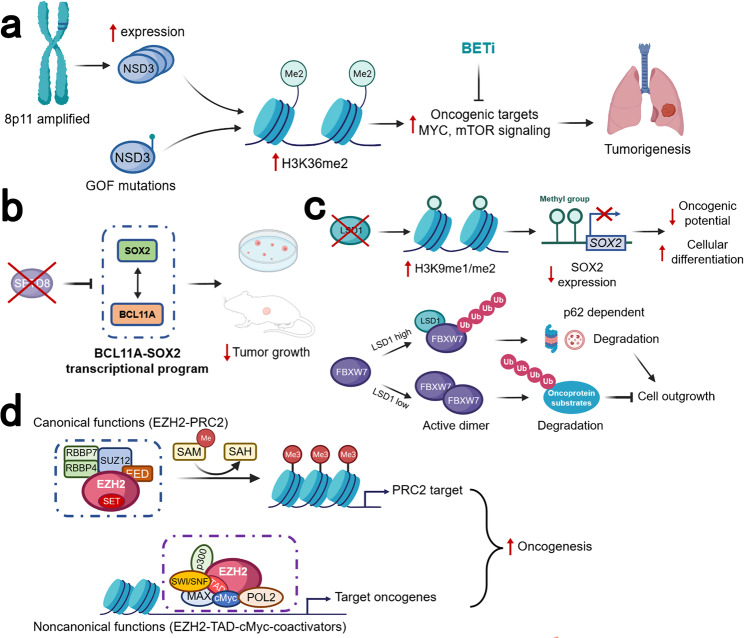
Lung cancer is the leading cause of cancer-related death across the world. Unlike lung adenocarcinoma, patients with lung squamous cell carcinoma (LSCC) have not benefitted from targeted therapies. Although immunotherapy has significantly improved cancer patients' outcomes, the relatively low response rate and severe adverse events hinder the clinical application of this promising treatment in LSCC. Therefore, it is of vital importance to have a better understanding of the mechanisms underlying the pathogenesis of LSCC as well as the inner connection among different signaling pathways, which will surely provide opportunities for more effective therapeutic interventions for LSCC. In this review, new insights were given about classical signaling pathways which have been proved in other cancer types but not in LSCC, including PI3K signaling pathway, VEGF/VEGFR signaling, and CDK4/6 pathway. Other signaling pathways which may have therapeutic potentials in LSCC were also discussed, including the FGFR1 pathway, EGFR pathway, and KEAP1/NRF2 pathway. Next, chromosome 3q, which harbors two key squamous differentiation markers SOX2 and TP63 is discussed as well as its related potential therapeutic targets. We also provided some progress of LSCC in epigenetic therapies and immune checkpoints blockade (ICB) therapies. Subsequently, we outlined some combination strategies of ICB therapies and other targeted therapies. Finally, prospects and challenges were given related to the exploration and application of novel therapeutic strategies for LSCC.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Landscape of targeted therapies for lung squamous cell carcinoma.Front Oncol. 2024 Oct 31;14:1467898. doi: 10.3389/fonc.2024.1467898. eCollection 2024. Front Oncol. 2024. PMID: 39544292 Free PMC article. Review.
-
Squamous cell lung cancer: Current landscape and future therapeutic options.Cancer Cell. 2022 Nov 14;40(11):1279-1293. doi: 10.1016/j.ccell.2022.09.018. Epub 2022 Oct 20. Cancer Cell. 2022. PMID: 36270277 Review.
-
Distinctive roles of syntaxin binding protein 4 and its action target, TP63, in lung squamous cell carcinoma: a theranostic study for the precision medicine.BMC Cancer. 2020 Sep 29;20(1):935. doi: 10.1186/s12885-020-07448-2. BMC Cancer. 2020. PMID: 32993587 Free PMC article.
-
Genomic insights into the mechanisms of FGFR1 dependency in squamous cell lung cancer.J Clin Invest. 2023 Nov 1;133(21):e174171. doi: 10.1172/JCI174171. J Clin Invest. 2023. PMID: 37909331 Free PMC article.
-
Targeting the cell signaling pathway Keap1-Nrf2 as a therapeutic strategy for adenocarcinomas of the lung.Expert Opin Ther Targets. 2019 Mar;23(3):241-250. doi: 10.1080/14728222.2019.1559824. Epub 2018 Dec 27. Expert Opin Ther Targets. 2019. PMID: 30556750 Review.
Cited by
-
Wee1 inhibitor PD0166285 sensitized TP53 mutant lung squamous cell carcinoma to cisplatin via STAT1.Cancer Cell Int. 2024 Sep 13;24(1):315. doi: 10.1186/s12935-024-03489-w. Cancer Cell Int. 2024. PMID: 39272147 Free PMC article.
-
Immunotherapy Improves the Survival of Stage 4 Non-Small Cell Lung Cancer Patients at the US Population Level: The Real-World Evidence.Clin Respir J. 2024 Sep;18(9):e70000. doi: 10.1111/crj.70000. Clin Respir J. 2024. PMID: 39275901 Free PMC article.
-
At the crossroads of immunotherapy for oncogene-addicted subsets of NSCLC.Nat Rev Clin Oncol. 2023 Mar;20(3):143-159. doi: 10.1038/s41571-022-00718-x. Epub 2023 Jan 13. Nat Rev Clin Oncol. 2023. PMID: 36639452 Review.
-
Characterization of platelet-related genes and constructing signature combined with immune-related genes for predicting outcomes and immunotherapy response in lung squamous cell carcinoma.Aging (Albany NY). 2023 Jul 20;15(14):6969-6992. doi: 10.18632/aging.204886. Epub 2023 Jul 20. Aging (Albany NY). 2023. PMID: 37477536 Free PMC article.
-
PHF12 regulates HDAC1 to promote tumorigenesis via EGFR/AKT signaling pathway in non-small cell lung cancer.J Transl Med. 2024 Jul 29;22(1):689. doi: 10.1186/s12967-024-05488-x. J Transl Med. 2024. PMID: 39075515 Free PMC article.
References
-
- Society, A. C. Cancer facts & figures 2022. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-... (2022).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
