

The epidermal growth factor receptor in healthy pregnancy and preeclampsia
- PMID: 36197759
- PMCID: PMC9742168
- DOI: 10.1530/JME-22-0105
The epidermal growth factor receptor in healthy pregnancy and preeclampsia
Abstract
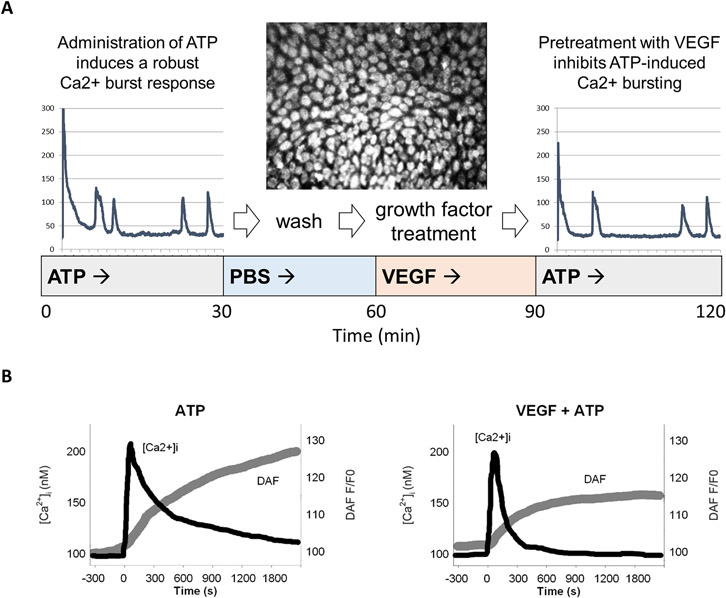
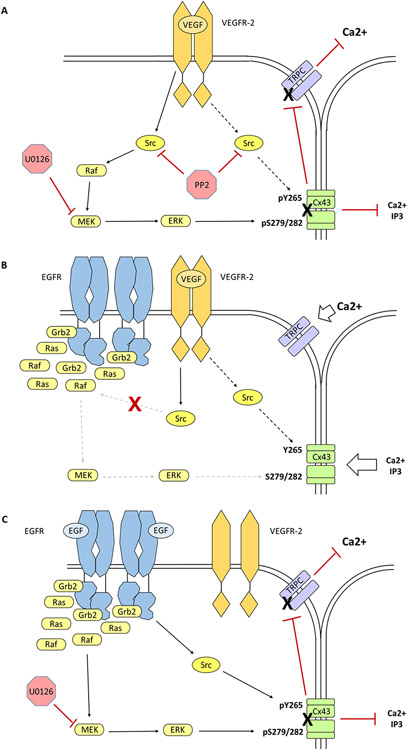
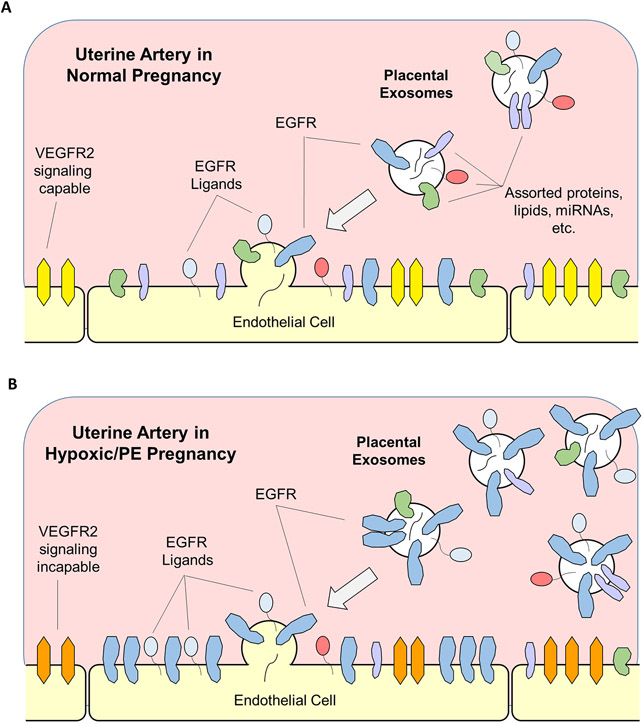
The epidermal growth factor receptor (EGFR) is expressed robustly in the placenta, and critical processes of pregnancy such as placental growth and trophoblast fusion are dependent on EGFR function. However, the role that aberrant EGFR signaling might play in the etiology and/or maintenance of preeclampsia (PE) remains largely unexplored. Recently, we have shown that overexpression of EGFR in cultured uterine artery endothelial cells (UAEC), which express little endogenous EGFR, remaps responsiveness away from vascular endothelial growth factor receptor (VEGFR) signaling and toward EGFR, suggesting that endothelial EGFR expression may be kept low to preserve VEGFR control of angiogenesis. Here we will consider the evidence for the possibility that the endothelial dysfunction observed in PE might in some cases result from elevation of endothelial EGFR. During pregnancy, trophoblasts are known to synthesize large amounts of EGFR protein, and the placenta regularly releases syncytiotrophoblast-derived exosomes and microparticles into the maternal circulation. Although there are no reports of elevated EGFR gene expression in preeclamptic endothelial cells, the ongoing shedding of placental vesicles into the vascular system raises the possibility that EGFR-rich vesicles might fuse with endothelium, thereby contributing to the symptoms of PE by interrupting angiogenesis and blocking pregnancy-adapted vasodilatory function.
Keywords: EGFR; endothelium; exosome; preeclampsia; pregnancy.
Conflict of interest statement
Declaration of Competing Interests
There are no conflicts of interest to disclose. Dr Ian Bird is a Senior Editor on the Journal of Endocrinology & Journal of Molecular Endocrinology join editorial board. Dr Ian Bird was not involved in the review or editorial process for this paper, on which he is listed as an author.
Figures
Similar articles
-
Adenoviral transduction of EGFR into pregnancy-adapted uterine artery endothelial cells remaps growth factor induction of endothelial dysfunction.Mol Cell Endocrinol. 2020 Jan 1;499:110590. doi: 10.1016/j.mce.2019.110590. Epub 2019 Sep 21. Mol Cell Endocrinol. 2020. PMID: 31550517 Free PMC article.
-
An integrated model of preeclampsia: a multifaceted syndrome of the maternal cardiovascular-placental-fetal array.Am J Obstet Gynecol. 2022 Feb;226(2S):S963-S972. doi: 10.1016/j.ajog.2020.10.023. Epub 2021 Mar 9. Am J Obstet Gynecol. 2022. PMID: 33712272 Review.
-
Placental-Specific sFLT-1 e15a Protein Is Increased in Preeclampsia, Antagonizes Vascular Endothelial Growth Factor Signaling, and Has Antiangiogenic Activity.Hypertension. 2015 Dec;66(6):1251-9. doi: 10.1161/HYPERTENSIONAHA.115.05883. Epub 2015 Sep 28. Hypertension. 2015. PMID: 26416849
-
EGFR (Epidermal Growth Factor Receptor) Signaling and the Mitochondria Regulate sFlt-1 (Soluble FMS-Like Tyrosine Kinase-1) Secretion.Hypertension. 2019 Mar;73(3):659-670. doi: 10.1161/HYPERTENSIONAHA.118.12300. Hypertension. 2019. PMID: 30636550
-
Expert review: preeclampsia Type I and Type II.Am J Obstet Gynecol MFM. 2023 Dec;5(12):101203. doi: 10.1016/j.ajogmf.2023.101203. Epub 2023 Oct 21. Am J Obstet Gynecol MFM. 2023. PMID: 37871693 Review.
Cited by
-
Rational Design of Nanomedicine for Placental Disorders: Birthing a New Era in Women's Reproductive Health.Small. 2024 Oct;20(41):e2300852. doi: 10.1002/smll.202300852. Epub 2023 May 16. Small. 2024. PMID: 37191231 Review.
-
Uterine macrophages and NK cells exhibit population and gene-level changes after implantation but maintain pro-invasive properties.Front Immunol. 2024 Mar 19;15:1364036. doi: 10.3389/fimmu.2024.1364036. eCollection 2024. Front Immunol. 2024. PMID: 38566989 Free PMC article.
-
Chemical mixture that targets the epidermal growth factor pathway impairs human trophoblast cell functions.Toxicol Appl Pharmacol. 2024 Feb;483:116804. doi: 10.1016/j.taap.2024.116804. Epub 2024 Jan 6. Toxicol Appl Pharmacol. 2024. PMID: 38185387 Free PMC article.
-
Profiling of MicroRNAs for the Identification of Unique and Common MicroRNAs in Preeclamptic Patients of South India Using Next-Generation Sequencing.Cureus. 2024 Oct 2;16(10):e70730. doi: 10.7759/cureus.70730. eCollection 2024 Oct. Cureus. 2024. PMID: 39493050 Free PMC article.
-
EGFR-targeted ionizable lipid nanoparticles enhance in vivo mRNA delivery to the placenta.J Control Release. 2024 Jul;371:455-469. doi: 10.1016/j.jconrel.2024.05.036. Epub 2024 Jun 10. J Control Release. 2024. PMID: 38789090
References
-
- Abeydeera LR, Wang WH, Cantley TC, Rieke A, Prather RS & Day BN 1998. Presence of epidermal growth factor during in vitro maturation of pig oocytes and embryo culture can modulate blastocyst development after in vitro fertilization. Mol Reprod Dev 51 395–401. - PubMed
-
- Argast GM, Campbell JS, Brooling JT & Fausto N 2004. Epidermal growth factor receptor transactivation mediates tumor necrosis factor-induced hepatocyte replication. J Biol Chem 279 34530–34536. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
