

A Boy with Sandestig-Stefanova Syndrome and Genital Abnormalities
- PMID: 36158057
- PMCID: PMC9421686
- DOI: 10.1159/000521331
A Boy with Sandestig-Stefanova Syndrome and Genital Abnormalities
Abstract
Introduction: Sandestig-Stefanova syndrome is an autosomal recessive developmental syndrome characterized by microcephaly, trigonocephaly, congenital cataracts, microphthalmia, facial findings, camptodactyly, periventricular white matter loss, thin corpus callosum, delayed myelination, and poor prognosis. This syndrome is caused by biallelic loss-of-function mutations in the NUP188 gene.
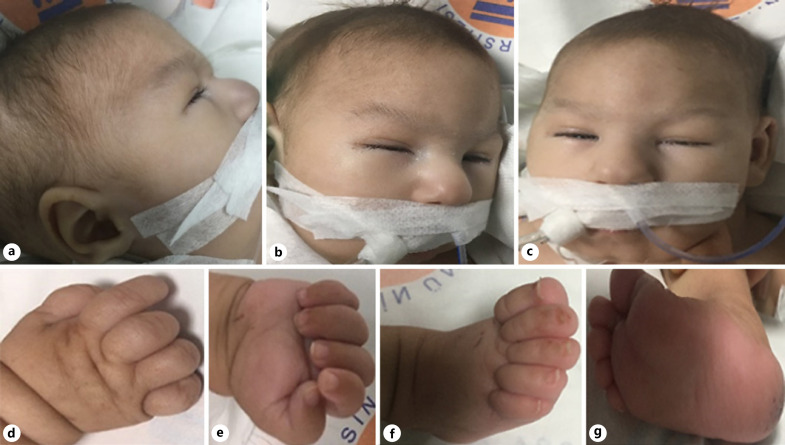
Case presentation: In the physical examination of our patient, whose mother and father were third-degree relatives, hypotonia, bilateral congenital cataracts, ambiguous genitalia, hypospadias, undescended testis, and facial dysmorphic findings (hypertelorism, high palate, micrognathia, microphthalmia, low-set ears) were detected.
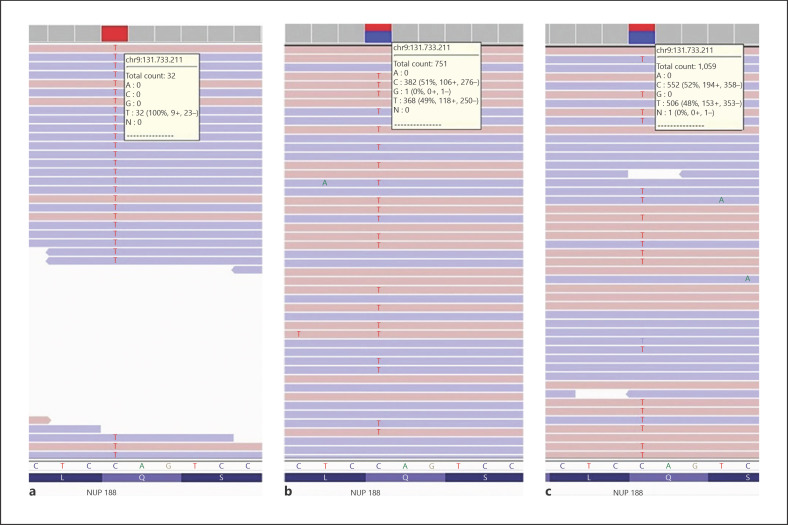
Discussion: In our patient, a homozygous c.1087C>T (p.Gln363Ter) variant was detected in exon 11 of the NUP188 (NM_015354.3) gene. The mother and father were found to be heterozygous carriers of this variant. All patients with the diagnosis of Sandestig-Stevanova syndrome reported in the literature are female. Our patient is the first male patient reported with this syndrome. In addition, immunodeficiency, congenital hypothyroidism, biotinidase deficiency, undescended testis, hypospadias, and ambiguous genitalia are defined for the first time in this syndrome. Our patient is the first case of Sandestig-Stefanova syndrome reported from Turkey. In this study, Sandestig-Stefanova syndrome with a novel pathogenic NUP188 gene variant is presented.
Keywords: Hypotonia; Microcephaly; Mutation; NUP188 gene; Sandestig-Stefanova syndrome.
Copyright © 2022 by S. Karger AG, Basel.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
NUP188 Biallelic Loss of Function May Underlie a New Syndrome: Nucleoporin 188 Insufficiency Syndrome?Mol Syndromol. 2020 Jan;10(6):313-319. doi: 10.1159/000504818. Epub 2019 Dec 10. Mol Syndromol. 2020. PMID: 32021605 Free PMC article.
-
Bi-allelic Loss-of-Function Variants in NUP188 Cause a Recognizable Syndrome Characterized by Neurologic, Ocular, and Cardiac Abnormalities.Am J Hum Genet. 2020 May 7;106(5):623-631. doi: 10.1016/j.ajhg.2020.03.009. Epub 2020 Apr 9. Am J Hum Genet. 2020. PMID: 32275884 Free PMC article.
-
Novel mutation in the RAB3GAP1 gene, the first diagnosed Warburg Micro syndrome case in Syria.Oxf Med Case Reports. 2020 May 23;2020(4):omaa031. doi: 10.1093/omcr/omaa031. eCollection 2020 Apr. Oxf Med Case Reports. 2020. PMID: 32477580 Free PMC article.
-
First Clinical Report of Two RAB3GAP1 Pathogenic Variant in Warburg Micro Syndrome.J Pediatr Genet. 2023 May 11;12(3):193-198. doi: 10.1055/s-0043-1768693. eCollection 2023 Sep. J Pediatr Genet. 2023. PMID: 37575647 Free PMC article. Review.
-
A case of Raine syndrome presenting with facial dysmorphy and review of literature.BMC Med Genet. 2018 May 11;19(1):76. doi: 10.1186/s12881-018-0593-x. BMC Med Genet. 2018. PMID: 29751744 Free PMC article. Review.
Cited by
-
The Expanding Phenotypic Spectrum of NUP188 Variants Points Toward Multiple Biological Pathways.Mol Syndromol. 2022 Jul;13(4):261-262. doi: 10.1159/000525275. Epub 2022 Jul 6. Mol Syndromol. 2022. PMID: 36158058 Free PMC article. No abstract available.
References
-
- Beck M, Hurt E. The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol. 2017;18:73–89. - PubMed
-
- Jühlen R, Fahrenkrog B. Moonlighting nuclear pore proteins: tissue-specific nucleoporin function in health and disease. Histochem Cell Biol. 2018;150((6)):593–605. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
