

Endogenous transcripts direct microRNA degradation in Drosophila, and this targeted degradation is required for proper embryonic development
- PMID: 36150386
- PMCID: PMC9648618
- DOI: 10.1016/j.molcel.2022.08.029
Endogenous transcripts direct microRNA degradation in Drosophila, and this targeted degradation is required for proper embryonic development
Abstract
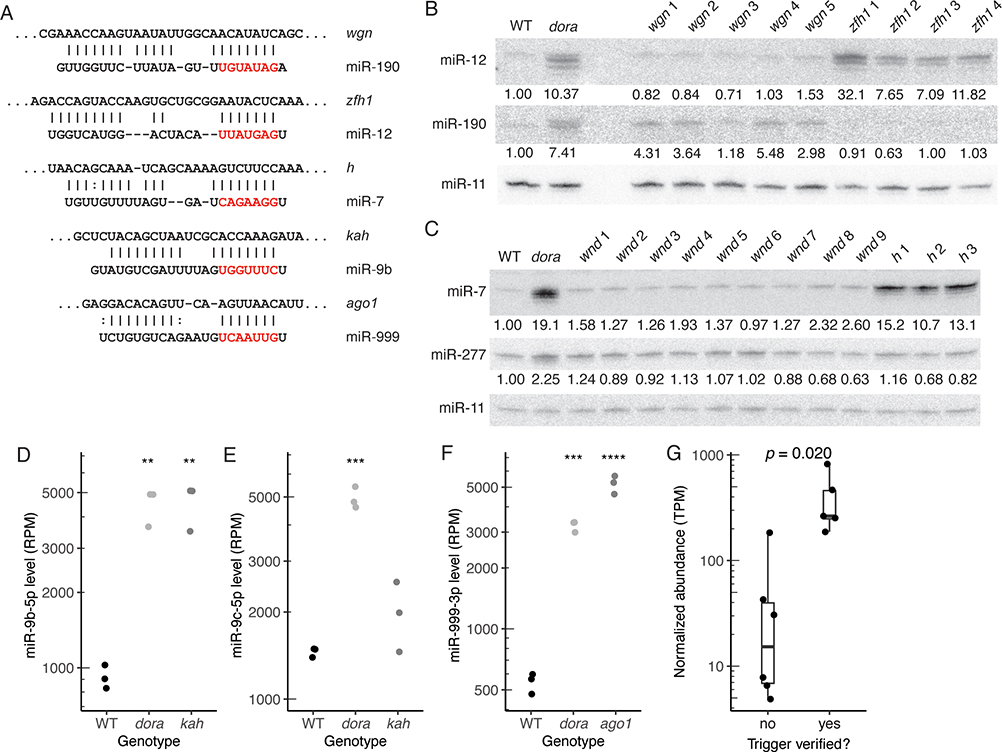
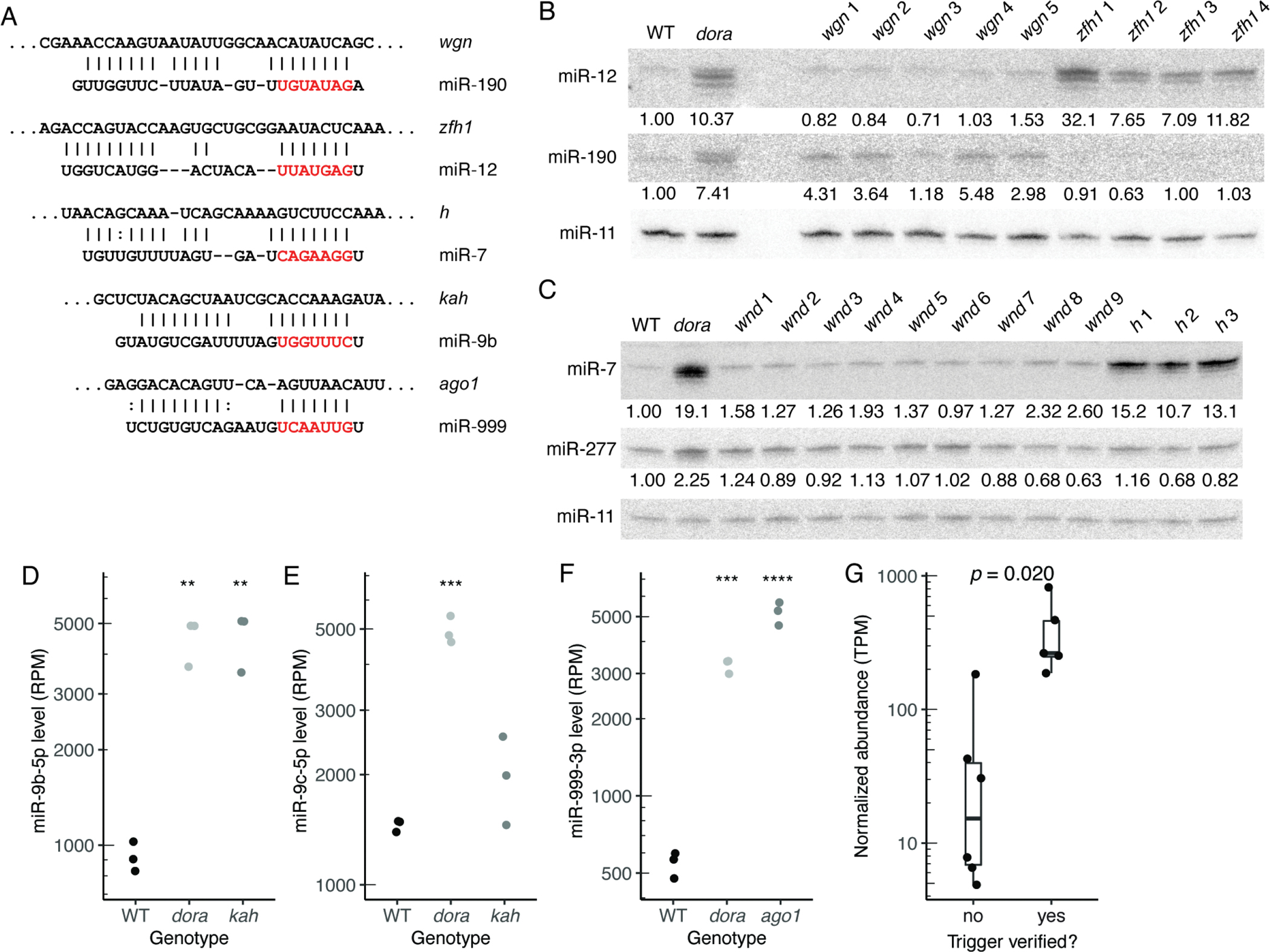
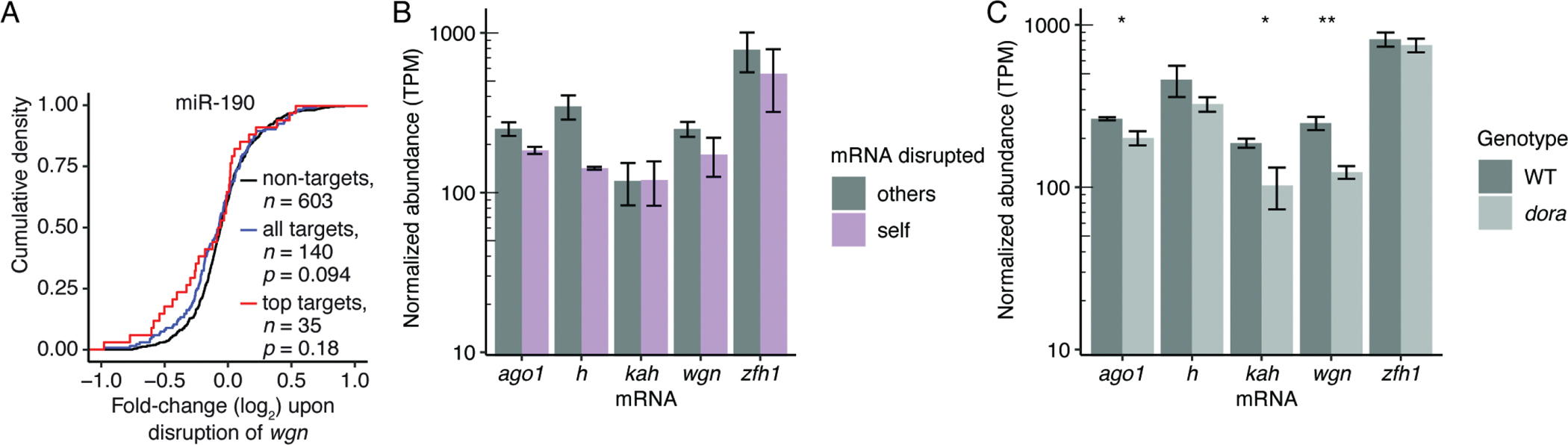
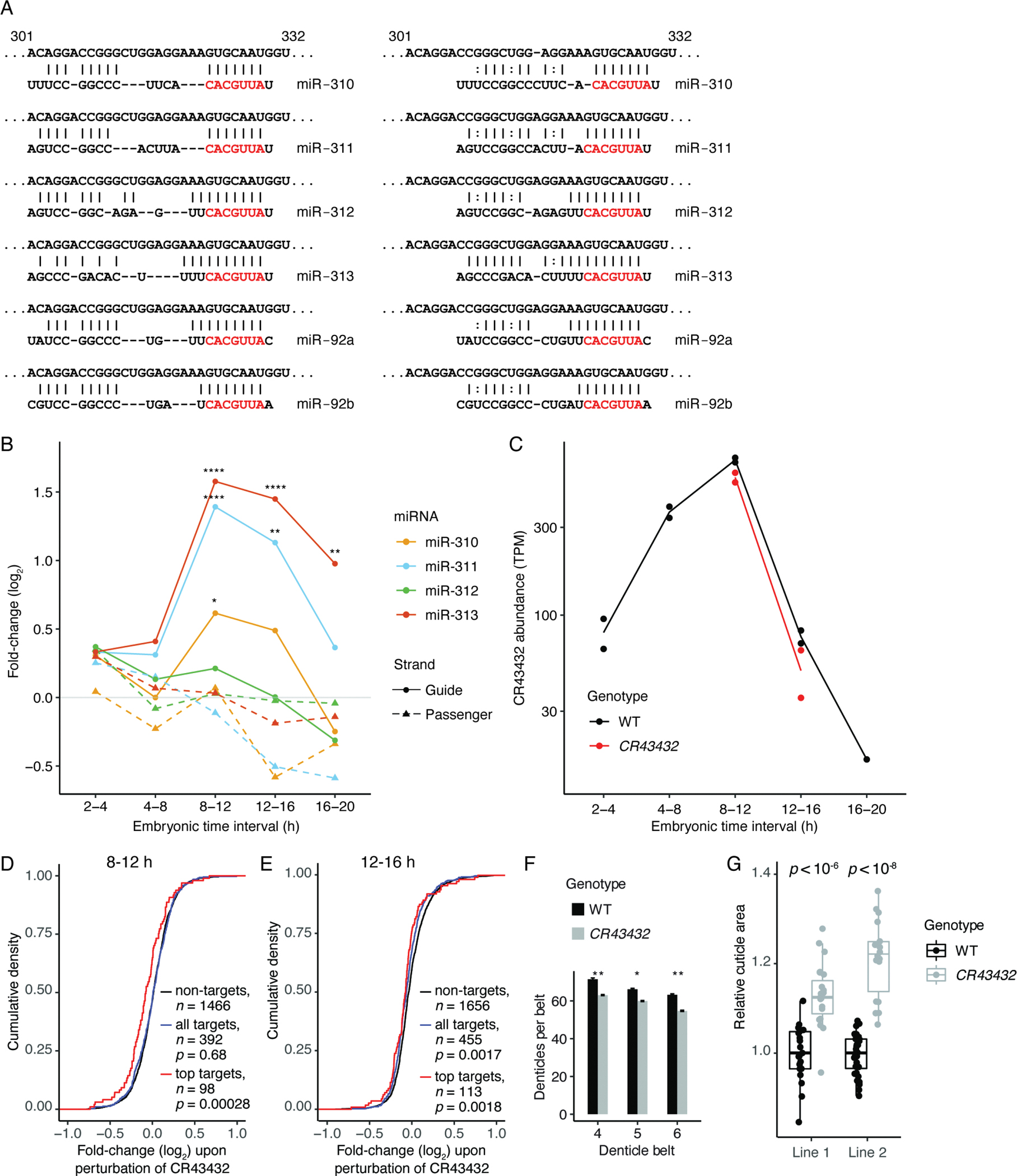
MicroRNAs (miRNAs) typically direct degradation of their mRNA targets. However, some targets have unusual miRNA-binding sites that direct degradation of cognate miRNAs. Although this target-directed miRNA degradation (TDMD) is thought to shape the levels of numerous miRNAs, relatively few sites that endogenously direct degradation have been identified. Here, we identify six sites, five in mRNAs and one in a noncoding RNA named Marge, which serve this purpose in Drosophila cells or embryos. These six sites direct miRNA degradation without collateral target degradation, helping explain the effectiveness of this miRNA-degradation pathway. Mutations that disrupt this pathway are lethal, with many flies dying as embryos. Concomitant derepression of miR-3 and its paralog miR-309 appears responsible for some of this lethality, whereas the loss of Marge-directed degradation of miR-310 miRNAs causes defects in embryonic cuticle development. Thus, TDMD is implicated in the viability of an animal and is required for its proper development.
Keywords: Drosophila embryonic development; Drosophila small RNAs; microRNAs; post-transcriptional gene regulation; target-directed miRNA degradation.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests D.P.B. is a member of Molecular Cell’s advisory board. E.R.K. has an immediate family member on Molecular Cell’s advisory board.
Figures
Similar articles
-
Target-directed microRNA degradation: Mechanisms, significance, and functional implications.Wiley Interdiscip Rev RNA. 2024 Mar-Apr;15(2):e1832. doi: 10.1002/wrna.1832. Wiley Interdiscip Rev RNA. 2024. PMID: 38448799 Free PMC article. Review.
-
Widespread microRNA degradation elements in target mRNAs can assist the encoded proteins.Genes Dev. 2021 Dec 1;35(23-24):1595-1609. doi: 10.1101/gad.348874.121. Epub 2021 Nov 24. Genes Dev. 2021. PMID: 34819352 Free PMC article.
-
Screening of Drosophila microRNA-degradation sequences reveals Argonaute1 mRNA's role in regulating miR-999.Nat Commun. 2023 Apr 13;14(1):2108. doi: 10.1038/s41467-023-37819-9. Nat Commun. 2023. PMID: 37055443 Free PMC article.
-
Target-directed microRNA degradation regulates developmental microRNA expression and embryonic growth in mammals.Genes Dev. 2023 Jul 1;37(13-14):661-674. doi: 10.1101/gad.350906.123. Epub 2023 Aug 8. Genes Dev. 2023. PMID: 37553261 Free PMC article.
-
MicroRNAs silence gene expression by repressing protein expression and/or by promoting mRNA decay.Cold Spring Harb Symp Quant Biol. 2006;71:523-30. doi: 10.1101/sqb.2006.71.013. Cold Spring Harb Symp Quant Biol. 2006. PMID: 17381335 Review.
Cited by
-
ZSWIM8 destabilizes many murine microRNAs and is required for proper embryonic growth and development.Genome Res. 2023 Sep;33(9):1482-1496. doi: 10.1101/gr.278073.123. Epub 2023 Aug 2. Genome Res. 2023. PMID: 37532519 Free PMC article.
-
Target-directed microRNA degradation: Mechanisms, significance, and functional implications.Wiley Interdiscip Rev RNA. 2024 Mar-Apr;15(2):e1832. doi: 10.1002/wrna.1832. Wiley Interdiscip Rev RNA. 2024. PMID: 38448799 Free PMC article. Review.
-
A conserved nutrient responsive axis mediates autophagic degradation of miRNA-mRNA hybrids in blood cell progenitors.Nucleic Acids Res. 2024 Jan 11;52(1):385-403. doi: 10.1093/nar/gkad1047. Nucleic Acids Res. 2024. PMID: 37994707 Free PMC article.
-
Target-directed microRNA degradation in Drosophila.Nat Rev Genet. 2022 Dec;23(12):713. doi: 10.1038/s41576-022-00544-y. Nat Rev Genet. 2022. PMID: 36207543 No abstract available.
-
Influence of RNA circularity on Target RNA-Directed MicroRNA Degradation.Nucleic Acids Res. 2024 Apr 12;52(6):3358-3374. doi: 10.1093/nar/gkae094. Nucleic Acids Res. 2024. PMID: 38381063 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
